
https://github.com/lh3/bioawk
1、基本思想
使用:
usage: bioawk [-F fs] [-v var=value] [-c fmt] [-tH] [-f progfile | 'prog'] [file ...]
bioawk基本思想是把组成不同类型的文件(sam、bam、fasta、fastq、vcf)的基本元素封装成变量,直接调用即可。
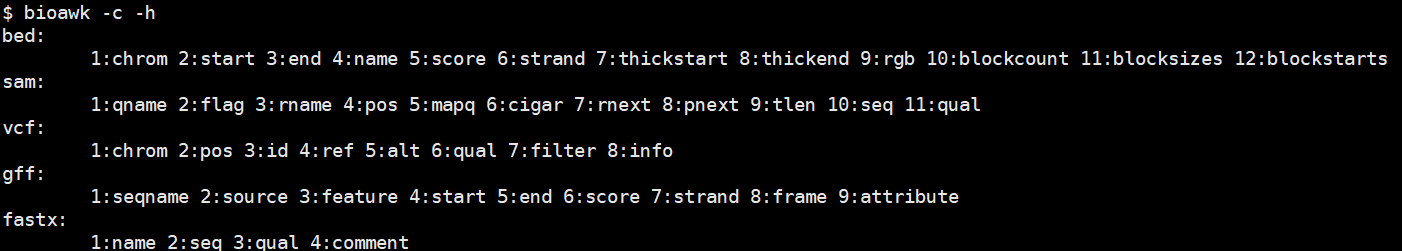
上面出现的名称即可引用其变量。
2、实际例子
打印fasta序列ID、序列、长度、GC含量:
bioawk -c fastx '{print "ID: "$name"\tlength: "length($seq)"\tGC: "gc($seq)"\t"$seq}' demo.fa
引用:https://blog.csdn.net/qq_42491125/article/details/92849378

 浙公网安备 33010602011771号
浙公网安备 33010602011771号