二次量子化与量子计算化学
 量子计算机,是由基本单元量子比特所组成的新型计算体系,通过量子叠加和量子纠缠的特性,来完成对量子态的操纵,最终再通过量子测量获得到想要的计算结果。而在量子计算机上面执行量子化学的任务,被认为是一个非常promising的应用场景,不论是从最初费曼的想法与设计,还是这几年所发展起来的近期量子计算(NISQ)的技术,都对量子计算化学这一新兴研究方向进行了阐述。本文通过最基础的谐振波,讲解到薛定谔方程和动量算符的由来,最终介绍了两种量子化的变换。其实所谓的量子化,都是对表征体系进行了调整。一次量子化将哈密顿量从电子的粒子性带到了量子力学中的波粒二象性,引入了动量算符。二次量子化将动量表象和位置表象变换到粒子数表象,通过统计平均的方法去研究电子在不同轨道之间跃迁时的能量吸收与产生,用于表示体系总能量。
量子计算机,是由基本单元量子比特所组成的新型计算体系,通过量子叠加和量子纠缠的特性,来完成对量子态的操纵,最终再通过量子测量获得到想要的计算结果。而在量子计算机上面执行量子化学的任务,被认为是一个非常promising的应用场景,不论是从最初费曼的想法与设计,还是这几年所发展起来的近期量子计算(NISQ)的技术,都对量子计算化学这一新兴研究方向进行了阐述。本文通过最基础的谐振波,讲解到薛定谔方程和动量算符的由来,最终介绍了两种量子化的变换。其实所谓的量子化,都是对表征体系进行了调整。一次量子化将哈密顿量从电子的粒子性带到了量子力学中的波粒二象性,引入了动量算符。二次量子化将动量表象和位置表象变换到粒子数表象,通过统计平均的方法去研究电子在不同轨道之间跃迁时的能量吸收与产生,用于表示体系总能量。
技术背景
二次量子化是量子化学(Quantum Chemistry)/量子计算化学(Quantum Computational Chemistry)中常用的一个模型,可以用于计算电子分布的本征能量和本征波函数。有一部分的物理学教材会认为二次量子化的这个叫法不大妥当,因为其本质是一种独立的正则变换,所以应该被称为第一种量子化(First Quantization)和第二种量子化(Second Quantization)。但是由于历史原因,就一直称呼为二次量子化。而如果认真去追究起来,称为二次量子化,可以理解为经历了两次的正则变换得到的结果,也并无不妥。本文将从比较原始的电子模型和启发式的薛定谔方程的推导讲起,尝试理解二次量子化发展过程中的各种物理图像。
一维无限长谐振子模型
在一个处于本征态的稳定的原子核-电子系统,如果把电子理解为一个粒子,那么所有的电子都处于围绕一个中心来回振动的状态,大致运动过程如下动态图(图片来自于参考链接1)所示:
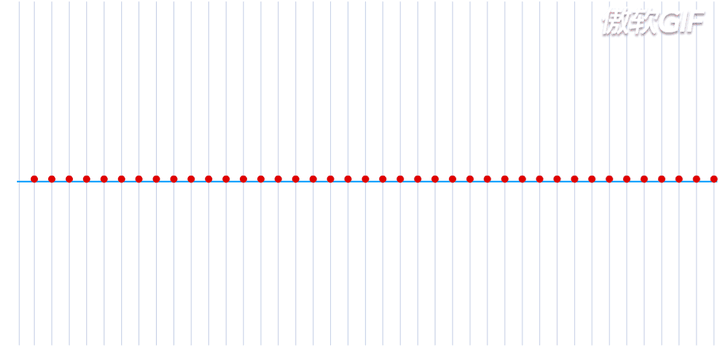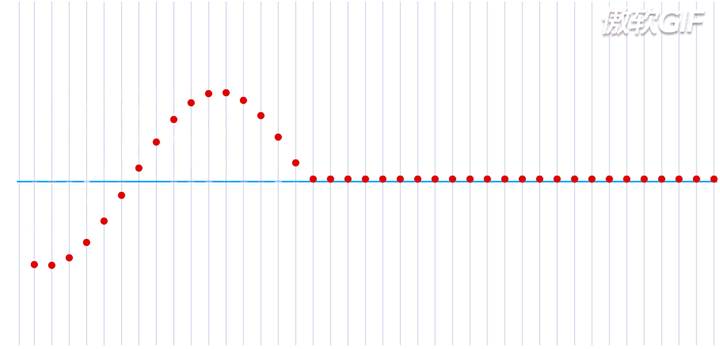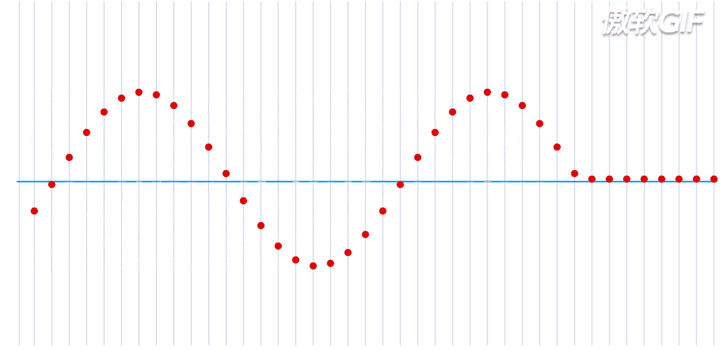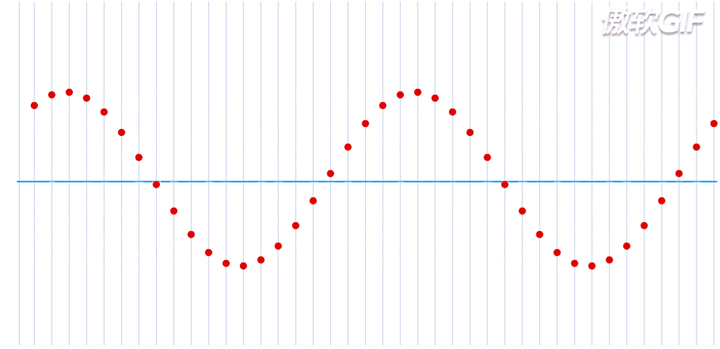
这就是一个平面简谐波,在横轴上进行传播,而给定一个时刻\(\tau\),又可以完整的描述其每一个位置所处的波形。如果给定一个位置进行观测,又可以得到任意时刻的振幅,即一维谐振子的运动方程:
完整的一维简谐波运动方程可以表示为:
之所以这里取了一个负号,是由于考虑了\(v=\frac{\partial y}{\partial t}\),即粒子运动速度的方向性。考虑完电子的粒子性,再考虑其波动性,我们不在把电子当做是在空间中来回运动的粒子,而是一个飘忽不定的“幽灵”。我们无法确定电子在空间中运动的轨迹,但是可以通过不断的测量,最终得到电子在空间中的一个分布图像,类似于下图的这种电子云的形式:

考虑波动性时,我们相当于在粒子性的基础之上施加两条约束:1. 得到的电子云的统计结果,要符合粒子性特征的密度分布,这就要保障实空间运动方程一致;2. 由于粒子性运动的范围有上下限,因此电子在粒子性运动空间内被观测到的总概率为1,也就是不可能在给定的运动空间之外检测到该电子。那么,基于波动性的要求,我们应当如此重写电子波函数:
如果是考虑三维的情况,把上式中的\(x\)替换为\(\bold{r}\)即可。
薛定谔方程的启发式推导
根据上一个章节中所给定的电子波函数,我们可以有如下的结论:
应用这些结论,我们可以逐一做推断,首先代入波函数的能量:\(E_{wave}=h\mu=\hbar\omega\)得到:
再考虑物质波的总能量:\(E_{wave}=\frac{p^2}{2m}+U\)和德布罗意波的动量表达式:\(p=\hbar k\),代入上式后得到:
其中拉普拉斯算子\(\nabla^2f=\sum^n_{i=1}\frac{\partial^2}{\partial x_i^2}f\),这样就完成了一个薛定谔方程的启发式推导。据说薛定谔最早就是这么想的,实际上这个推导过程有太多的限制条件,并不是一个通用的推导,但是推导出来的结果,也就是定态薛定谔方程,在各种实验结果下都得到了很好的验证。
量子力学算符
在上面关于动量的式子中,我们经过整理之后可以得到:
这里之所以把动量写成了\(\hat{p}\),是因为其不再是一个表征具体系统特性的一个量,而是一个量子力学的操作算符。得到动量算符的表达式之后,我们可以计算这样的一对对易子:
这个推论称之为正则对易关系,很多量子力学算符都有类似的特性,如果从矩阵的角度来理解,就是两个不满足交换律的矩阵的乘法。在上一个章节中,如果去掉两边的波函数,我们发现等式右边虽然形式发生了变化,但实际上还是表征体系总能量的特性,即:
这是哈密顿算符的由来,关于哈密顿算符的更多特性,这里先不展开介绍。但是我们可以根据非算符形式的哈密顿量,回顾一下先前的博客中介绍过的拉格朗日力学和哈密顿力学,我们可以计算得:
此即哈密顿量在动量和坐标表象下的哈密顿正则方程。
通用电子结构问题与BO近似
对于一个有\(K\)个原子核与\(N\)个电子的系统而言,其系统哈密顿量(总能量)可以表示为:
其中大写的代表原子核的参数,小写的代表电子的参数。为了简化模型,我们引入玻恩-奥本海默近似(Born-Oppenheimer approximation,简称BO近似):
通俗的说就是,由于原子核与电子的质量不在同一个量级,可以近似的将系统波函数看做是原子核波函数与电子波函数的乘积。而通常我们在量子化学中所考虑的问题都是给定的构象,也就是固定了原子核的位置,则原子核之间的势能可以看做是一个常数。这就使得,我们可以只关注电子这一块的哈密顿量与波函数,简化后得到的哈密顿量形式如下:
一次量子化
上一个章节中所得到的电子哈密顿量,只能用于表征其粒子性,为了同时满足电子的波粒二象性,我们需要对其参量进行正则变换。但是第一步我们可以仅简单的对变量(动量表象和位置表象)做等价变换:直接将动量算符\(\hat{p}=-i\hbar\frac{\partial}{\partial x}\)代入其中。而位置表象虽然从绝对位置表象\(r_i\)变换到了相对位置表象\(r_i-r_j\),但是我们可以发现其偏导数保持不变,则维持了哈密顿正则方程的不变性,是一个正则变换。最终我们得到的一次量子化(或者叫第一类量子化,First Quantization)的电子哈密顿量结果为:
需要注意的是,这里同时做了无量纲化:坐标取波尔长度,能量取Hartree能量。一次量子化的波函数,一般通过Slater行列式来表示:
其中\(M\)表示电子轨道数(其实每一个电子轨道都可以理解成一个能级),\(\phi_0(\bold{x_1})\)所表示的物理含义是第1个电子处于第0个电子轨道。这种波函数表征形式的好处在于,保证了每一个轨道上最多只有一个电子(电子的自旋上下被划分为两个电子轨道)。而且由于行列式的特性,Slater行列式还具有反演对称性(任意交换两个电子所处的位置,会使得符号取反):
但是看这个哈密顿量和波函数的形式我们就知道,如果我们想处理这种连续空间的变量是比较困难的,要尽可能的将其转换成离散化的变量,因此就产生了第二种量子化的方法。
二次量子化
二次量子化(Second Quantization),也称为第二种量子化,将动量与相对位置表象变换到了离散化的粒子数表象,通过产生算符\(a^{\dagger}\)和湮灭算符\(a\)来控制给定能级/电子轨道的粒子数(0或1)。按照这种方法,我们可以将哈密顿量写为如下的单体相互作用和两体相互作用形式:
其中单体相互作用参数和两体相互作用参数分别为:
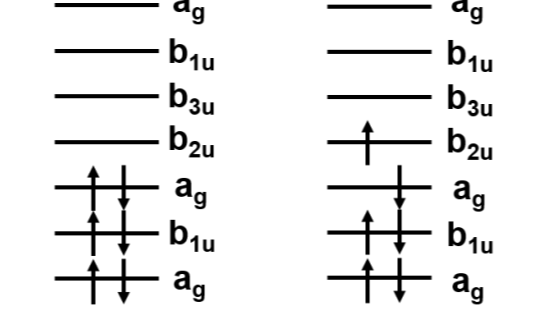
这两个参数分别所表征的物理含义为:电子从q轨道跃迁到p轨道的动能和原子核势能增减总和,在紧束缚模型下又称为hopping energy(电子跳跃能),以及两个电子分别从r、s轨道跃迁到q、p轨道的电子之间相互作用势能的变化(单体动能和原子核势能在前面一项中已经考虑到了)。关于对位置的积分,可以理解为给定的轨道占据一定的空间,空间上的任一位置的跃迁都被认为是该轨道所发生的跃迁,因此要遍历轨道所占据的空间。如下图所示是一个电子跃迁示意(图片来自于参考链接3):
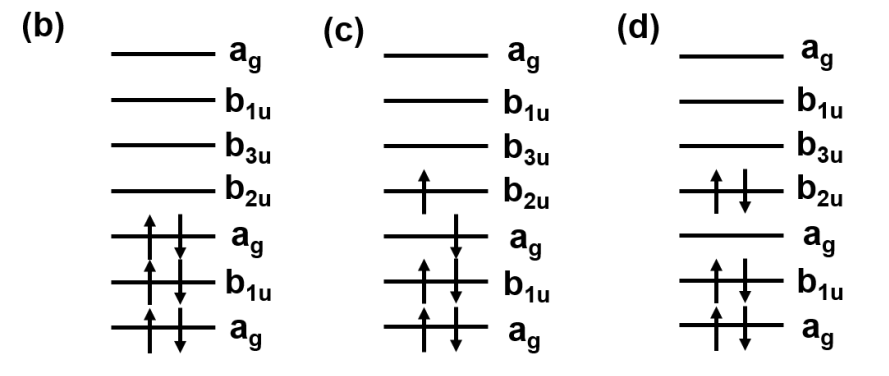
如此一来,我们就可以将一个量子比特定义成一个电子轨道,从而实现在量子计算机上面完成量子化学计算的任务。
总结概要
量子计算机,是由基本单元量子比特所组成的新型计算体系,通过量子叠加和量子纠缠的特性,来完成对量子态的操纵,最终再通过量子测量获得到想要的计算结果。而在量子计算机上面执行量子化学的任务,被认为是一个非常promising的应用场景,不论是从最初费曼的想法与设计,还是这几年所发展起来的近期量子计算(NISQ)的技术,都对量子计算化学这一新兴研究方向进行了阐述。本文通过最基础的谐振波,讲解到薛定谔方程和动量算符的由来,最终介绍了两种量子化的变换。其实所谓的量子化,都是对表征体系进行了调整。一次量子化将哈密顿量从电子的粒子性带到了量子力学中的波粒二象性,引入了动量算符。二次量子化将动量表象和位置表象变换到粒子数表象,通过统计平均的方法去研究电子在不同轨道之间跃迁时的能量吸收与产生,用于表示体系总能量。
版权声明
本文首发链接为:https://www.cnblogs.com/dechinphy/p/second-quantization.html
作者ID:DechinPhy
更多原著文章请参考:https://www.cnblogs.com/dechinphy/
打赏专用链接:https://www.cnblogs.com/dechinphy/gallery/image/379634.html
腾讯云专栏同步:https://cloud.tencent.com/developer/column/91958
参考链接
- https://zhuanlan.zhihu.com/p/139133715
- Quantum Computational Chemistry. Sam McArdle, Suguru Endo and other co-authors.
- https://arxiv.org/abs/2109.02110v1

 浙公网安备 33010602011771号
浙公网安备 33010602011771号