蛋白质基础组成结构
 本文通过对Xponge+VMD的工具对蛋白质进行建模,然后总结了20种氨基酸的具体信息,也就是蛋白质的基本组成单元。通过对这些氨基酸的组合,就可以得到一个具有生物活性的蛋白质。同时本文还介绍了常用的存储蛋白质结构的文件格式pdb的具体格式化定义,总体来说是一个总结性的文章。
本文通过对Xponge+VMD的工具对蛋白质进行建模,然后总结了20种氨基酸的具体信息,也就是蛋白质的基本组成单元。通过对这些氨基酸的组合,就可以得到一个具有生物活性的蛋白质。同时本文还介绍了常用的存储蛋白质结构的文件格式pdb的具体格式化定义,总体来说是一个总结性的文章。
技术背景
了解蛋白质的基本组成单元和结构,有助于了解蛋白质的特性。对于蛋白质结构的研究,在医药领域是非常核心的重要工作。这里我们仅仅介绍一些蛋白质的基本组成单元——20种氨基酸的种类,以及可以用于蛋白质建模的一些工具。
Xponge的安装和使用
Xponge是一款基于python开发的可以用于蛋白质建模的软件,可以用pip进行安装和管理:
$ python3 -m pip install xponge --upgrade
Requirement already satisfied: xponge in /home/dechin/anaconda3/envs/mindspore16/lib/python3.9/site-packages (1.2.5.9)
Requirement already satisfied: NetCDF4 in /home/dechin/anaconda3/envs/mindspore16/lib/python3.9/site-packages (from xponge) (1.5.8)
Requirement already satisfied: pubchempy in /home/dechin/anaconda3/envs/mindspore16/lib/python3.9/site-packages (from xponge) (1.0.4)
Requirement already satisfied: numpy in /home/dechin/anaconda3/envs/mindspore16/lib/python3.9/site-packages (from xponge) (1.20.3)
Requirement already satisfied: cftime in /home/dechin/anaconda3/envs/mindspore16/lib/python3.9/site-packages (from NetCDF4->xponge) (1.6.0)
使用的方法是在python文件或者python终端窗口中导入xponge和相关力场之后直接调用相关接口:
$ python3
Python 3.9.12 (main, Apr 5 2022, 06:56:58)
[GCC 7.5.0] :: Anaconda, Inc. on linux
Type "help", "copyright", "credits" or "license" for more information.
>>> import Xponge
>>> import Xponge.forcefield.AMBER.ff14SB
Reference for ff14SB:
James A. Maier, Carmenza Martinez, Koushik Kasavajhala, Lauren Wickstrom, Kevin E. Hauser, and Carlos Simmerling
ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB
Journal of Chemical Theory and Computation 2015 11 (8), 3696-3713
DOI: 10.1021/acs.jctc.5b00255
>>> Save_PDB(ALA,'ALA.pdb') # 内置了所有的氨基酸种类,注意开头结尾的氨基酸名称有所变化
>>> exit()
执行完Save之后,就会在当前的路径下生成一个ALA.pdb的文件:
$ cat ALA.pdb
REMARK Generated By Xponge (Molecule)
ATOM 1 N ALA 1 -3.801 -7.214 0.158
ATOM 2 H ALA 1 -2.791 -7.214 0.158
ATOM 3 CA ALA 1 -4.487 -5.938 0.158
ATOM 4 HA ALA 1 -5.112 -5.861 -0.732
ATOM 5 CB ALA 1 -5.374 -5.792 1.390
ATOM 6 HB1 ALA 1 -4.761 -5.858 2.289
ATOM 7 HB2 ALA 1 -5.877 -4.826 1.363
ATOM 8 HB3 ALA 1 -6.118 -6.588 1.399
ATOM 9 C ALA 1 -3.497 -4.783 0.158
ATOM 10 O ALA 1 -2.287 -4.998 0.158
可以用VMD之类的软件打开pdb文件,看一下其分子结构:
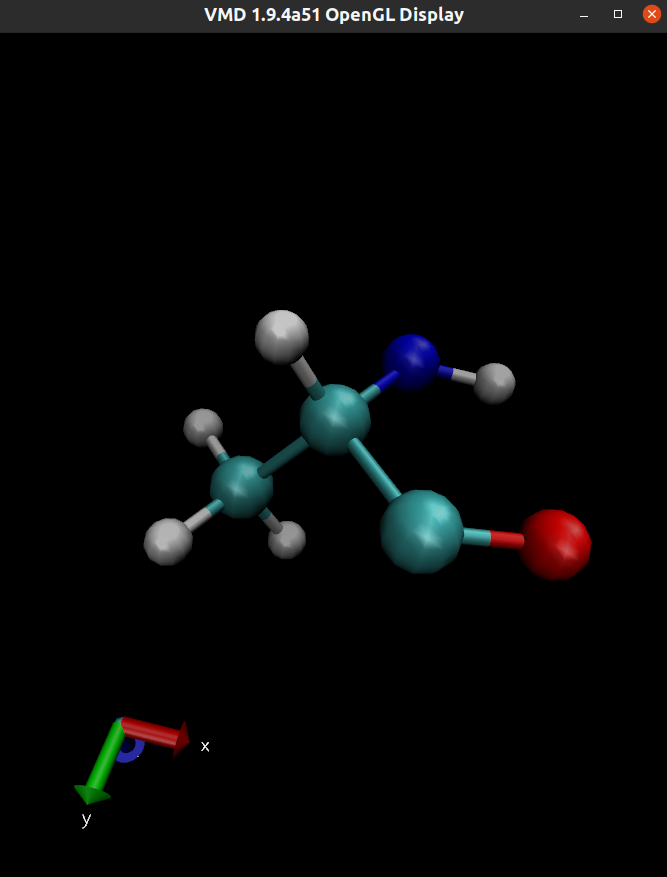
这里面需要注意的是,使用Xponge生成的氨基酸是以及去掉了一个\(H_2O\)的,分别是开头与\(N\)相连的\(H\)和结尾与\(C\)相连的\(OH\)。这种操作的合理之处在于,我们很少关注单个氨基酸的作用,如果要组装成一个蛋白质的模型,生成肽键的过程会脱水,因此在构建氨基酸的时候直接去掉\(H_2O\)可以在一定程度上提升计算的性能。另外就是,除了ACE和NME之外,氨基酸放在开头和结尾处时,分别要加上N和C作为标识。比如,当ALA放在蛋白质chain的开头时,其名称应为NALA,以此类推。还有一个值得提醒的点是,组氨酸本身包含了多种结构,在使用时需要指定特定的结构名称,否则默认就是HIS。
类似的方法,我们还可以用Xponge建立一个更加大型的模型:
In [1]: import Xponge
In [2]: import Xponge.forcefield.AMBER.ff14SB
Reference for ff14SB.py:
James A. Maier, Carmenza Martinez, Koushik Kasavajhala, Lauren Wickstrom, Kevin E. Hauser, and Carlos Simmerling
ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB
Journal of Chemical Theory and Computation 2015 11 (8), 3696-3713
DOI: 10.1021/acs.jctc.5b00255
In [3]: Save_PDB(ACE+ALA*10+NME,'multi_ALA.pdb')
生成的pdb文件,用vmd打开之后显示如下:

使用还是非常的方便。
氨基酸种类
这里是从参考链接2中整理出来的数据,以及用xponge和vmd画出来的三维结构图,主要是总结记录一下这些基本的组成单元。
| 英文名 | 中文名 | 三字母缩写 | 单字母缩写 | 结构式 | 等电点pI | 三维结构图 |
|---|---|---|---|---|---|---|
| Alanine | 丙氨酸 | Ala | A | CH3-CH(NH2)-COOH | 6.0 |  |
| Arginine | 精氨酸 | Arg | R | HN=C(NH2)-NH-(CH2)3-CH(NH2)-COOH | 10.76 |  |
| Asparagine | 天冬酰胺 | Asn | N | H2N-CO-CH2-CH(NH2)-COOH | 5.41 |  |
| Asparticacid | 天冬氨酸 | Asp | D | HOOC-CH2-CH(NH2)-COOH | 2.77 |  |
| Cysteine | 半胱氨酸 | Cys | C | HS-CH2-CH(NH2)-COOH | 5.05 |  |
| Glutamine | 谷氨酰胺 | Gln | Q | H2N-CO-(CH2)2-CH(NH2)-COOH | 5.41 |  |
| Glutamicacid | 谷氨酸 | Glu | E | HOOC-(CH2)2-CH(NH2)-COOH | 3.22 |  |
| Glycine | 甘氨酸 | Gly | G | NH2-CH2-COOH | 5.97 |  |
| Histidine | 组氨酸 | His | H | NH-CH=N-CH=C-CH2-CH(NH2)-COOH | 7.59 |  |
| Isoleucine | 异亮氨酸 | Ile | I | CH3-CH2-CH(CH3)-CH(NH2)-COOH | 6.02 |  |
| Leucine | 亮氨酸 | Leu | L | (CH3)2CH-CH2-CH(NH2)-COOH | 5.98 |  |
| Lysine | 赖氨酸 | Lys | K | H2N-(CH2)4-CH(NH2)-COOH | 9.74 |  |
| Methionine | 甲硫氨酸(蛋氨酸) | Met | M | CH3-S-(CH2)2-CH(NH2)-COOH | 5.74 |  |
| Phenylalanine | 苯丙氨酸 | Phe | F | Ph-CH2-CH(NH2)-COOH | 5.48 |  |
| Proline | 脯氨酸 | Pro | P | NH-(CH2)3-CH-COOH | 6.30 |  |
| Serine | 丝氨酸 | Ser | S | HO-CH2-CH(NH2)-COOH | 5.68 |  |
| Threonine | 苏氨酸 | Thr | T | CH3-CH(OH)-CH(NH2)-COOH | 6.16 |  |
| Tryptophan | 色氨酸 | Trp | W | Ph-NH-CH=C-CH2-CH(NH2)-COOH | 5.89 |  |
| Tyrosine | 酪氨酸 | Tyr | Y | HO-p-Ph-CH2-CH(NH2)-COOH | 5.66 |  |
| Valine | 缬氨酸 | Val | V | (CH3)2CH-CH(NH2)-COOH | 5.96 |  |
PDB文件基本格式
pdb是最常用的一种存储蛋白质结构的文本文件格式,但是pdb本身又是一个严格的结构化的文本文件,其对应位置的内容为:
| 列 | 数据 | 格式, 对齐 | 说明 |
|---|---|---|---|
| 1-4 | ATOM | 字符, 左 | Record Type 记录类型 |
| 7-11 | serial | 整数, 右 | Atom serial number 原子序号.PDB文件对分子结构处理为segment, chain, residue, atom四个层次(一般并不用到chain), 因此此数位限定了一个残基中的最大原子数为为99999 |
| 13-16 | name | 字符, 左 | Atom name 原子名称.原子的元素符号在13-14列中右对齐一般从14列开始写, 占四个字符的原子名称才会从13列开始写.如, 铁原子FE写在13-14列, 而碳原子C只写在14列. |
| 17 | altLoc | 字符 | Alternate location indicator 可替位置标示符 |
| 18-20 | resName | 字符 | Residue name 残基名称 |
| 22 | chainID | 字符 | Chain identifier 链标识符 |
| 23-26 | resSeq | 整数, 右 | Residue sequence number 残基序列号 |
| 27 | iCode | 字符 | Code for insertion of residues 残基插入码 |
| 28-30 | 留空 | ||
| 31-38 | x | 浮点, 右 | Orthogonal coordinates for X in Angstroms 直角x坐标(埃) |
| 39-46 | y | 浮点, 右 | Orthogonal coordinates for Y in Angstroms 直角y坐标(埃) |
| 47-54 | z | 浮点, 右 | Orthogonal coordinates for Z in Angstroms 直角z坐标(埃) |
| 55-60 | occupancy | 浮点, 右 | Occupancy 占有率 |
| 61-66 | tempFactor | 浮点, 右 | Temperature factor 温度因子 |
| 67-72 | 留空 | ||
| 73-76 | segID | 字符, 左 | Segment identifier(optional) 可选的片段标识符, VMD会使用此数据 |
| 77-78 | element | 字符, 右 | Element symbol 元素符号 |
| 79-80 | charge | 字符 | Charge on the atom(optional) 可选的原子电荷. 实际分子模拟中往往重新定义电荷, 故此列往往不用. VMD写出的PDB文件中无此列. |
这个格式里面有一个比较坑的点是,atom_name占位符长度会影响对齐位置。为了方便操作,这里用一个python的脚本来写pdb文件,也可以作为理解上述结构化参数的出发点:
def write_pdb(crd, atom_names, res_names, res_ids, pdb_name='temp.pdb'):
"""Write protein crd information into pdb format files.
Args:
crd(numpy.float32): The coordinates of protein atoms.
atom_names(numpy.str_): The atom names differ from aminos.
res_names(numpy.str_): The residue names of amino names.
res_ids(numpy.int32): A unique mask each same residue.
pdb_name(str): The path to save the pdb file, absolute path is suggested.
"""
success = 1
with open(pdb_name, 'w') as pdb:
pdb.write('MODEL 1\n')
for i,c in enumerate(crd[0]):
pdb.write('ATOM'.ljust(6))
pdb.write('{}'.format(i + 1).rjust(5))
if len(atom_names[i])<4:
pdb.write(' ')
pdb.write(atom_names[i].ljust(3))
else:
pdb.write(' ')
pdb.write(atom_names[i].ljust(4))
pdb.write(res_names[i].rjust(4))
pdb.write('A'.rjust(2))
pdb.write('{}'.format(res_ids[i]).rjust(4))
pdb.write(' ')
pdb.write('{:.3f}'.format(c[0]).rjust(8))
pdb.write('{:.3f}'.format(c[1]).rjust(8))
pdb.write('{:.3f}'.format(c[2]).rjust(8))
pdb.write('1.0'.rjust(6))
pdb.write('0.0'.rjust(6))
pdb.write('{}'.format(atom_names[i][0]).rjust(12))
pdb.write('\n')
pdb.write('TER\n')
pdb.write('ENDMDL\n')
pdb.write('END\n')
return success
这样只要给定crd(原子坐标),atom_names(原子名称),res_names(残基/氨基酸名称),res_ids(残基位置编号)这几个参数,就可以生成一个符合格式要求的pdb文件。如果不严格按照这个pdb格式写入,有可能存在其他工具无法读取的情况,比如上一篇博客中所介绍的TMscore软件就要求非常严格。
总结概要
本文通过对Xponge+VMD的工具对蛋白质进行建模,然后总结了20种氨基酸的具体信息,也就是蛋白质的基本组成单元。通过对这些氨基酸的组合,就可以得到一个具有生物活性的蛋白质。同时本文还介绍了常用的存储蛋白质结构的文件格式pdb的具体格式化定义,总体来说是一个总结性的文章。
版权声明
本文首发链接为:https://www.cnblogs.com/dechinphy/p/pdb.html
作者ID:DechinPhy
更多原著文章请参考:https://www.cnblogs.com/dechinphy/
打赏专用链接:https://www.cnblogs.com/dechinphy/gallery/image/379634.html
腾讯云专栏同步:https://cloud.tencent.com/developer/column/91958
CSDN同步链接:https://blog.csdn.net/baidu_37157624?spm=1008.2028.3001.5343
51CTO同步链接:https://blog.51cto.com/u_15561675

 浙公网安备 33010602011771号
浙公网安备 33010602011771号