
MindSponge分子动力学模拟——自建力场(2024.03)
 基于力场的分子动力学模拟,其实可以看做是一个最简单的机器学习模型,具有计算成本低的特点,在药物研发、生物化学和计算物理学等研究领域存在广泛的应用。那么,如何去快速的开发一个新的力场,在传统的MD模拟软件中其实可能是一个不小的门槛,而基于MindSpore框架开发的MindSponge分子动力学模拟软件,则具有这种便捷开发的特性。本文通过一个简单的示例,介绍了如何在MindSponge分子动力学模拟框架内构建一个自定义的分子力场,可以正常的执行分子动力学模拟迭代过程并保存相应的结果和输出。
基于力场的分子动力学模拟,其实可以看做是一个最简单的机器学习模型,具有计算成本低的特点,在药物研发、生物化学和计算物理学等研究领域存在广泛的应用。那么,如何去快速的开发一个新的力场,在传统的MD模拟软件中其实可能是一个不小的门槛,而基于MindSpore框架开发的MindSponge分子动力学模拟软件,则具有这种便捷开发的特性。本文通过一个简单的示例,介绍了如何在MindSponge分子动力学模拟框架内构建一个自定义的分子力场,可以正常的执行分子动力学模拟迭代过程并保存相应的结果和输出。
技术背景
在MindSponge教程合集中我们已经介绍了很多使用MindSponge进行分子动力学模拟的方法,这里主要介绍在MindSponge中自定义一个力场。在传统的MD软件中,如果你希望去开发一个自己的力场,或者是添加一些分子动力学模拟方法如增强采样等,会面临不少编程上的困难。而这些困难对于使用Python来编程的MindSponge来说,就天然的降低了门槛。以力场为例子,我们可以在EnergyCell的基础上,去开发一个自定义的ForceField。
自定义力场
首先,关于MindSponge的安装和基础使用,大家可以在前面的博客中去学习,这里不做更多介绍。
由于这里我们只是为了演示如何使用MindSponge来构建自定义力场的方法,因此我们可以用一个最简单方便的操作,把所有的坐标做一个加和:
这里\(E_{self}\)就是我们用到的自定义力场的势能。在MindSponge里面定义一个力场,首先我们继承基础能量类EnergyCell来构建一个新的势能类:
class MyEnergy(EnergyCell):
def construct(self, coordinate: Tensor, **kwargs):
return coordinate.sum()[None, None]
这里需要解释的是,因为我们只是定义一个简单的力场,没有额外的参数,所以没必要修改__init__函数里面的参数内容,只需要修改construct里面的内容就可以了。唯一有要求的是最后输出的能量的维度有一点要求,为了原生支持多batch的操作,我们输出的能量项是二维的,所以这里求和之后又做了一个扩维。完整的演示代码如下所示:
from mindspore import context, Tensor
from mindspore.nn import Adam
context.set_context(mode=context.GRAPH_MODE, device_target='GPU')
if __name__ == "__main__":
from sponge import Sponge, Molecule, WithEnergyCell
from sponge.potential import EnergyCell, ForceFieldBase
from sponge.callback import RunInfo, SaveLastPdb
# 自定义能量类
class MyEnergy(EnergyCell):
def construct(self, coordinate: Tensor, **kwargs):
return coordinate.sum()[None, None]
# 生成水分子系统
system = Molecule(template='water.spce.yaml')
system.reduplicate([0.3, 0, 0])
new_sys = system.copy([0, 0, -0.3])
system.append(new_sys)
# 根据系统生成自定义力场对象
potential = MyEnergy(system)
forcefield = ForceFieldBase(potential)
withenergy = WithEnergyCell(system, forcefield)
opt = Adam(system.trainable_params(), 1e-3)
mini = Sponge(withenergy, optimizer=opt)
# 配置回调函数
run_info = RunInfo(5)
save_pdb = SaveLastPdb(system, save_freq=1, pdb_name='water_last.pdb')
# 开始执行模拟任务
mini.run(10, callbacks=[run_info, save_pdb])
这里演示的是4个水分子的体系,其中定义好能量项之后,要用ForceFieldBase力场基础类封装起来,才是一个完整的力场。然后再把力场信息、系统信息和迭代器信息传给Sponge类,就可以来时进行模拟了。根据定义的迭代器的不同,既可以完成能量优化的功能,也可以实现分子动力学模拟的过程,在框架上实现了统一。此外我们还可以分别定义两个回调函数RunInfo和SaveLastPdb,用于在屏幕上输出迭代信息,以及输出最后一步的系统的坐标到一个指定的pdb文件里面。运行结果如下所示:
[MindSPONGE] Started simulation at 2024-03-22 16:54:45
[MindSPONGE] Step: 5, E_pot: 0.31788814
[MindSPONGE] Step: 10, E_pot: 0.13788882
[MindSPONGE] Finished simulation at 2024-03-22 16:54:46
[MindSPONGE] Simulation time: 1.03 seconds.
--------------------------------------------------------------------------------
同时在当前路径下生成了一个pdb文件:
MODEL 1
ATOM 1 O WAT A 1 -0.100 -0.100 -0.100 1.0 0.0 O
ATOM 2 H1 WAT A 1 0.716 0.477 -0.100 1.0 0.0 H
ATOM 3 H2 WAT A 1 -0.916 0.477 -0.100 1.0 0.0 H
ATOM 4 O WAT A 2 2.900 -0.100 -0.100 1.0 0.0 O
ATOM 5 H1 WAT A 2 3.716 0.477 -0.100 1.0 0.0 H
ATOM 6 H2 WAT A 2 2.084 0.477 -0.100 1.0 0.0 H
ATOM 7 O WAT A 3 -0.100 -0.100 -3.100 1.0 0.0 O
ATOM 8 H1 WAT A 3 0.716 0.477 -3.100 1.0 0.0 H
ATOM 9 H2 WAT A 3 -0.916 0.477 -3.100 1.0 0.0 H
ATOM 10 O WAT A 4 2.900 -0.100 -3.100 1.0 0.0 O
ATOM 11 H1 WAT A 4 3.716 0.477 -3.100 1.0 0.0 H
ATOM 12 H2 WAT A 4 2.084 0.477 -3.100 1.0 0.0 H
TER
ENDMDL
END
有了这个pdb结构文件,我们就可以用VMD来进行可视化了:
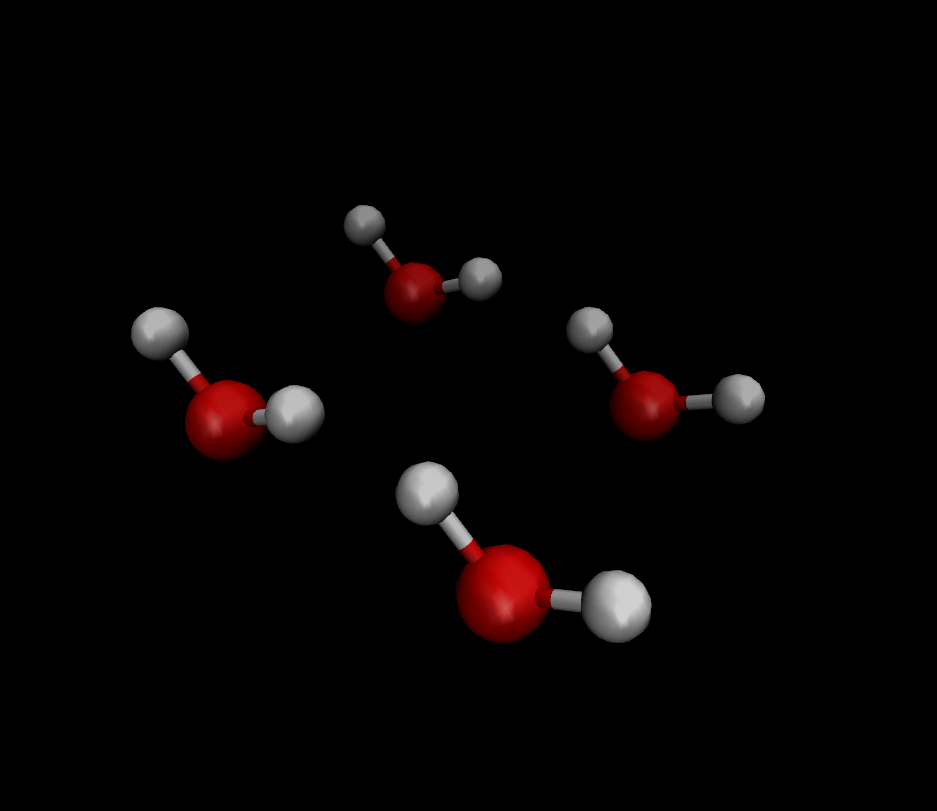
如果需要保存完整的轨迹文件,那就需要用到MindSponge所支持的h5md数据结构,相关方法可以参考这篇博客中的范例,其中还包含了使用MDAnalysis第三方软件进行后分析的方法。
总结概要
基于力场的分子动力学模拟,其实可以看做是一个最简单的机器学习模型,具有计算成本低的特点,在药物研发、生物化学和计算物理学等研究领域存在广泛的应用。那么,如何去快速的开发一个新的力场,在传统的MD模拟软件中其实可能是一个不小的门槛,而基于MindSpore框架开发的MindSponge分子动力学模拟软件,则具有这种便捷开发的特性。本文通过一个简单的示例,介绍了如何在MindSponge分子动力学模拟框架内构建一个自定义的分子力场,可以正常的执行分子动力学模拟迭代过程并保存相应的结果和输出。
版权声明
本文首发链接为:https://www.cnblogs.com/dechinphy/p/energy-cell.html
作者ID:DechinPhy
更多原著文章:https://www.cnblogs.com/dechinphy/
请博主喝咖啡:https://www.cnblogs.com/dechinphy/gallery/image/379634.html

