

MestReNova图谱分析
一、MestReNove基础设置及便捷操作
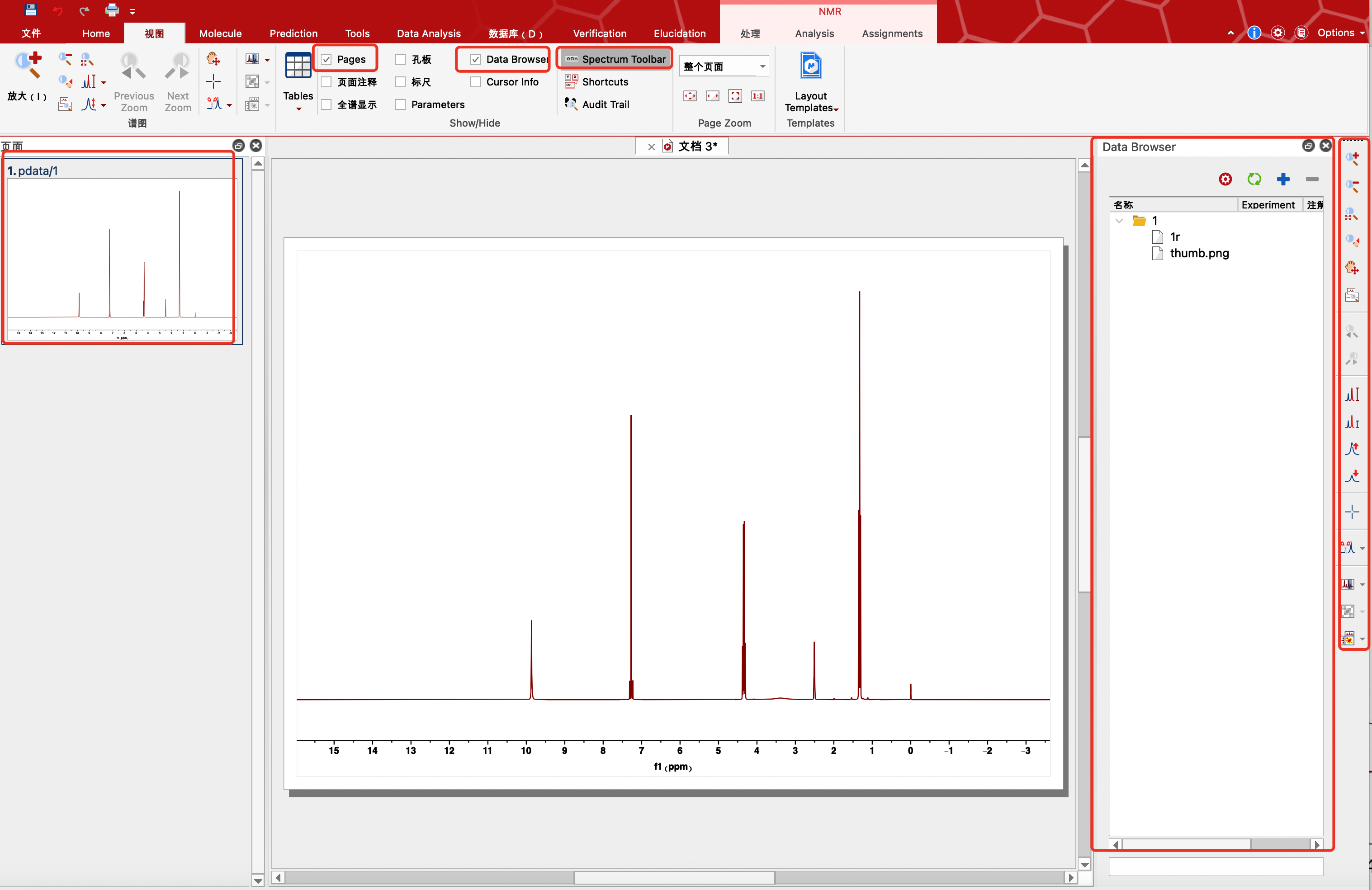
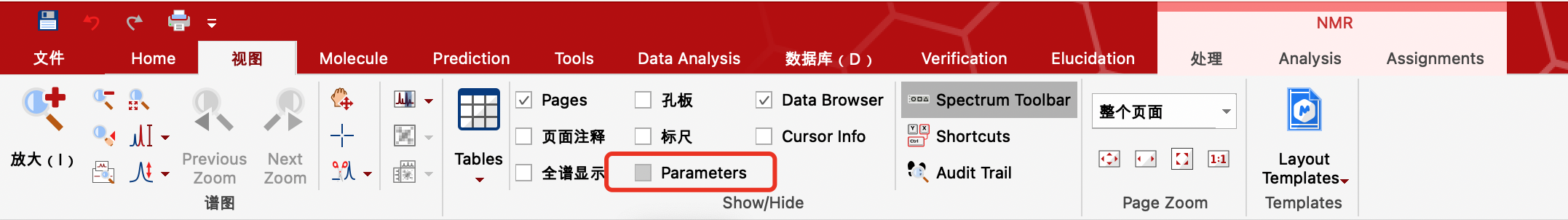
view(视图)中
勾选Pages显示左侧页面。
勾选Data Browser显示文件库,可移至右侧,导入文件,方便查看。一般都是打开1r文件即可。
点击Shortcuts可查看快捷键。
勾选Spectrum Toolbar(打开1r文件之后),右侧显示基本操作,里面包含放大缩小,全谱,移动等功能,可以尝试使用快捷键简便操作。其中放大功能可以从左向右滑动放大选定区域。
鼠标放在图谱空白区域滑动滚轮,图谱的纵坐标放大或缩小。
二、NMR图谱操作
2.1 图谱预处理
在processing(处理)中进行基线校正和相位校正。
(1)图谱放大后基线不平时可以进行基线校正
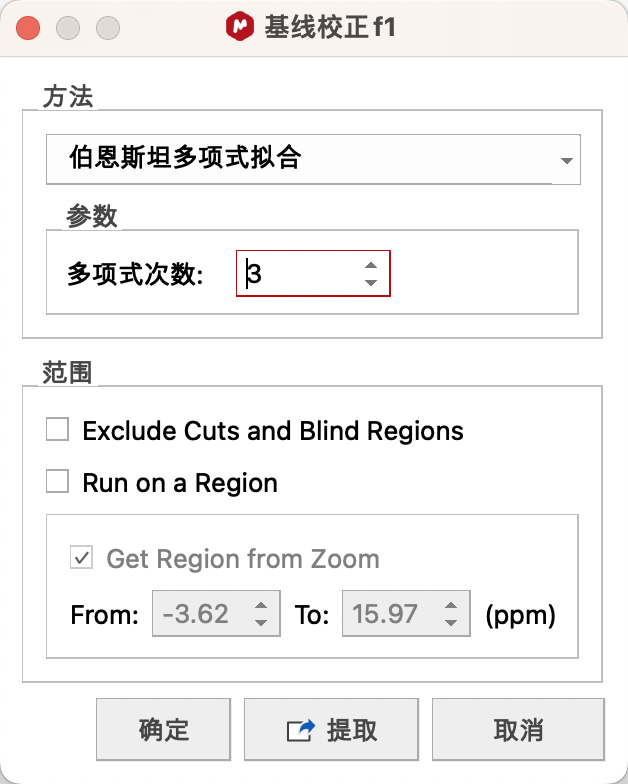
Auto Baseline Correction——自动基线校正
基础设置:点击Auto Baseline Correction处下拉菜单中的Baseline Correction…(基线校正…)
方法推荐Bernstein Polynomial Fit 拟合方法(伯恩斯坦多项式拟合,第三个)。
点击图标或者全自动(Full Auto )将自动按照设置的方法进行校正。
(2)若图形相位不好,左右扭曲,可以进行相位校正
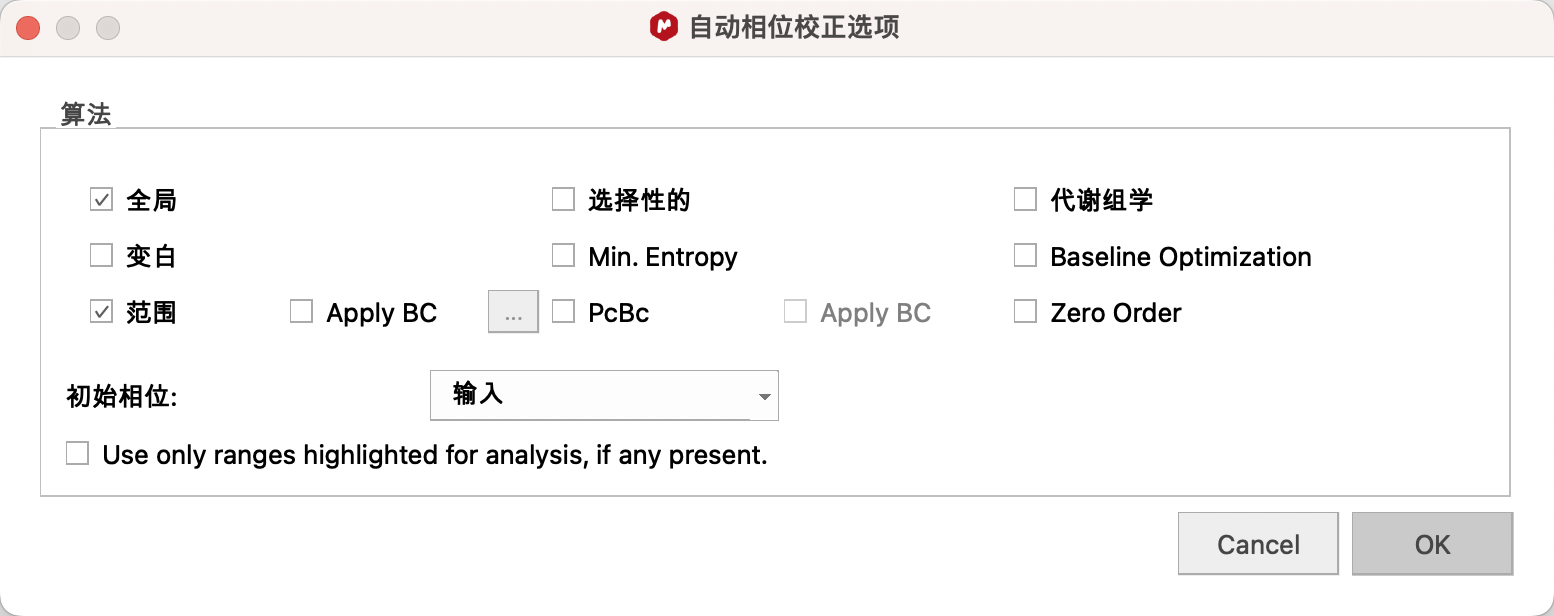
1、Auto Phase Correction——自动相位校正(建议使用)
基础设置:点击Auto Phase Correction处下拉菜单中的Options(选项)。
针对一维氢谱,建议勾选G lobal(全局),Regions(范围),取消勾选Selective(选择性的,碳谱谱图可勾选 )。
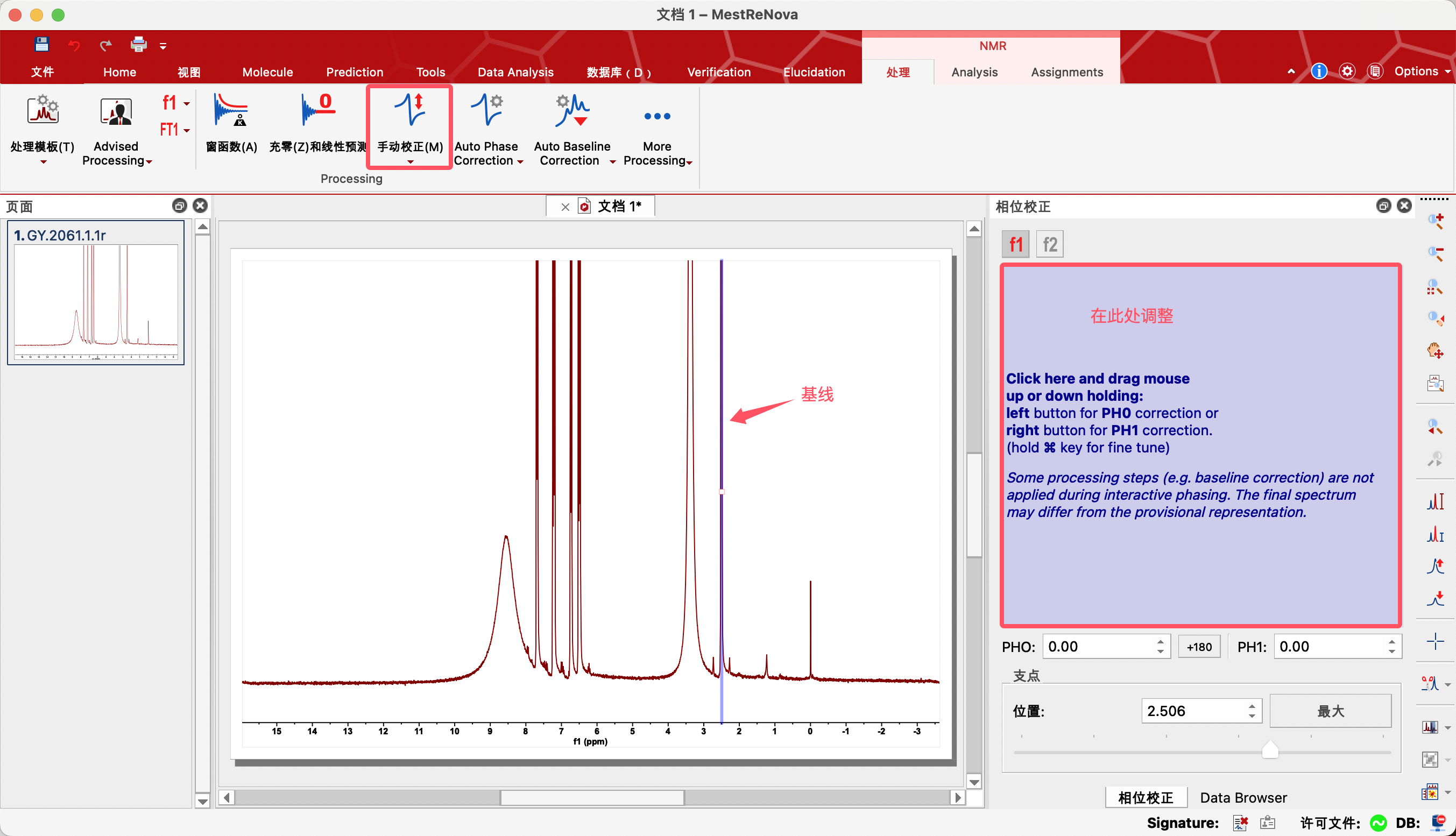
2、Manual Correction——手动相位校正
针对自动相位校正不理想的情况下,可以进行手动相位校正。
点击手动校正——点击右侧蓝色区域,会在最高最主要的一个信号上面出现一条浅蓝色的竖线,作为一个支点——将鼠标放在蓝色区域,按着鼠标的左键不动,上下移动鼠标,使蓝色竖线左右相位对称,对于其他距离蓝色竖线较远的地方,按着鼠标的右键不动,上下移动鼠标进行调整。调整结果仍不好时,可以移动蓝色竖线以其他信号作为支点进行调整。
2.2 图谱分析
在Analysis(分析)中进行定标,标识和积分。
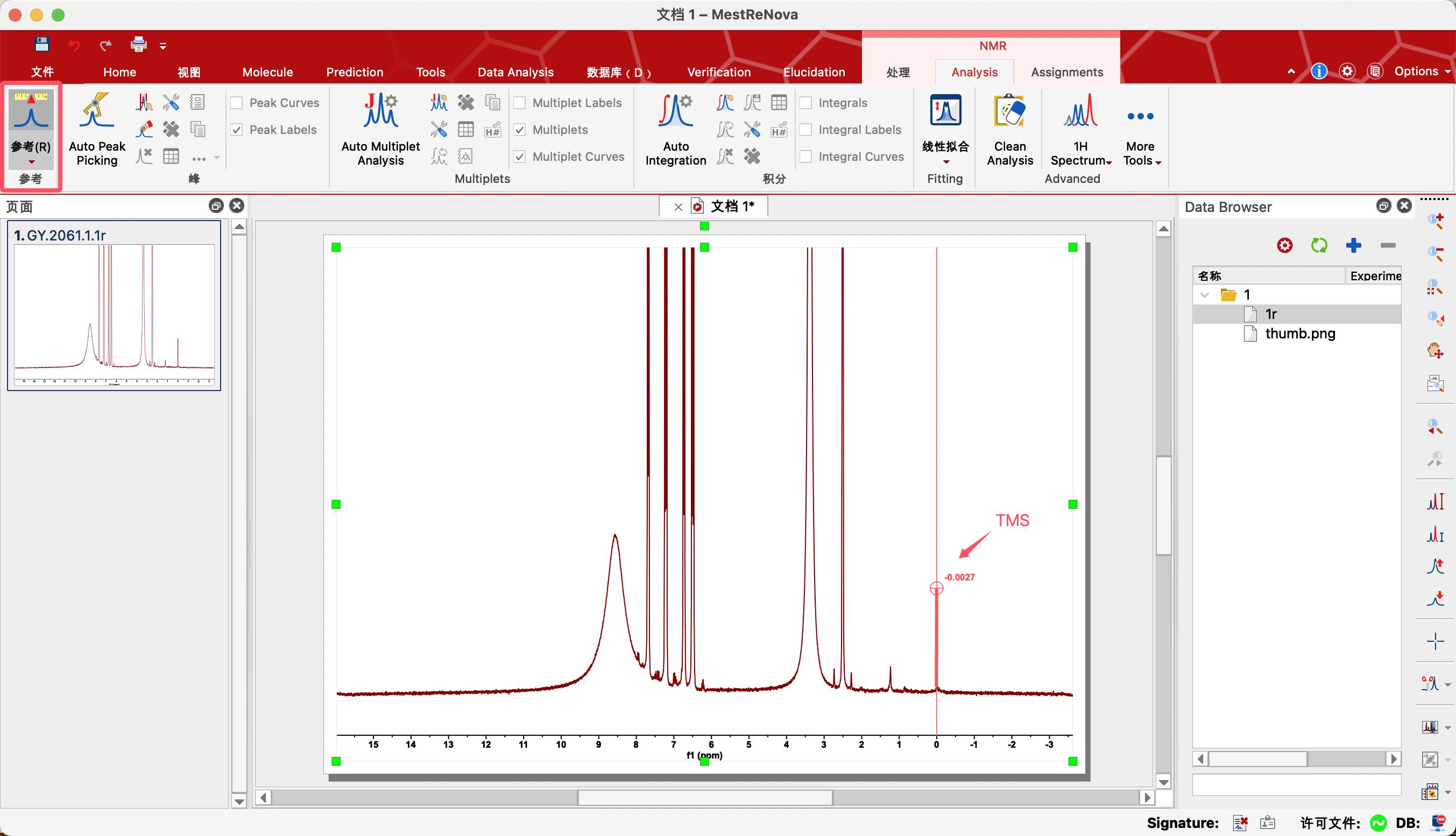
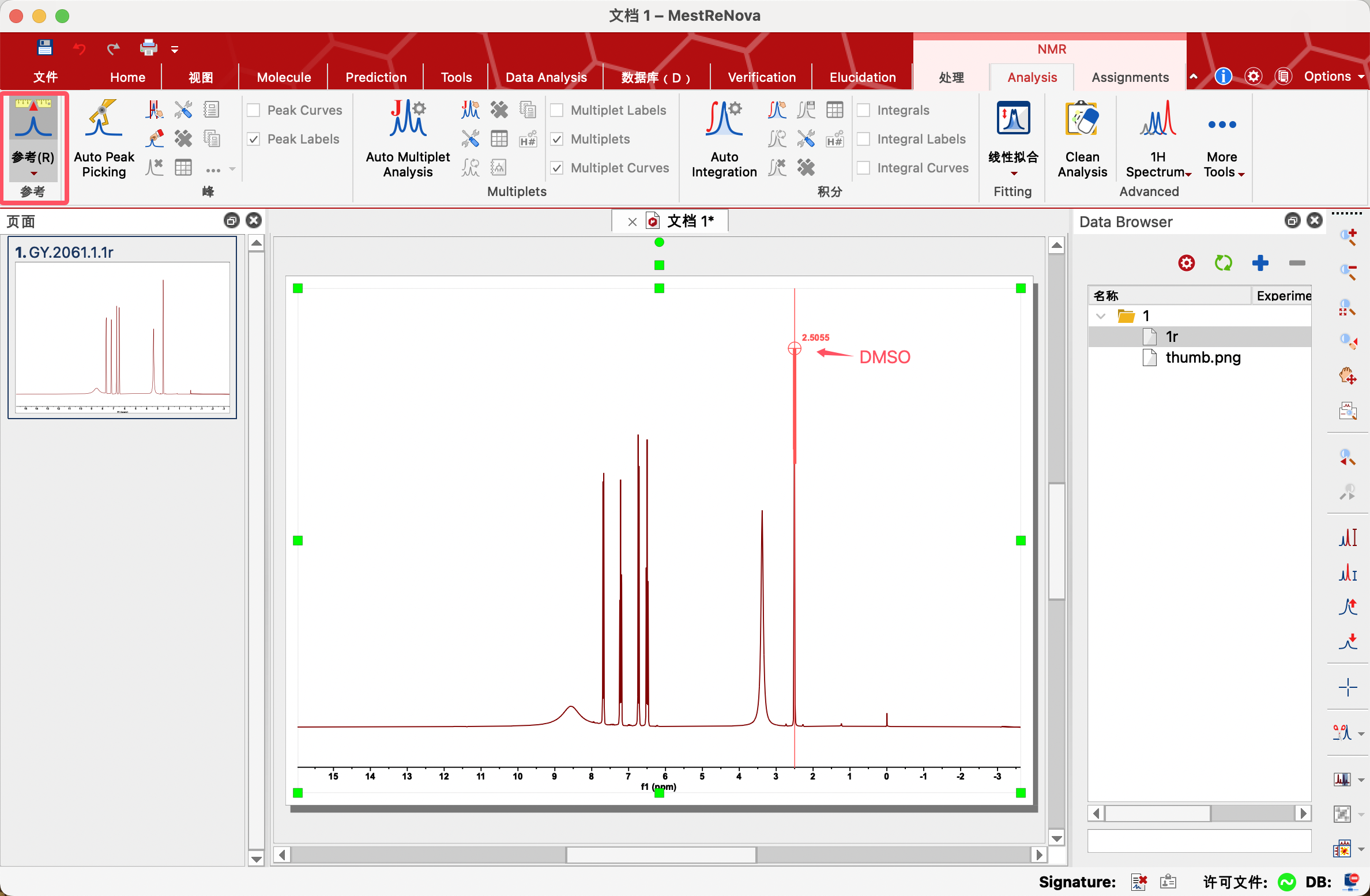
(1)步骤一:Reference(参考)——定标
对于一张氢谱,通常情况下是需要进行定标的。一般使用TMS或者其他溶剂进行定标。
查看谱图的溶剂型号:在View(视图)中勾选Parameters,Parameters窗口中的Solvent项目显示溶剂型号。
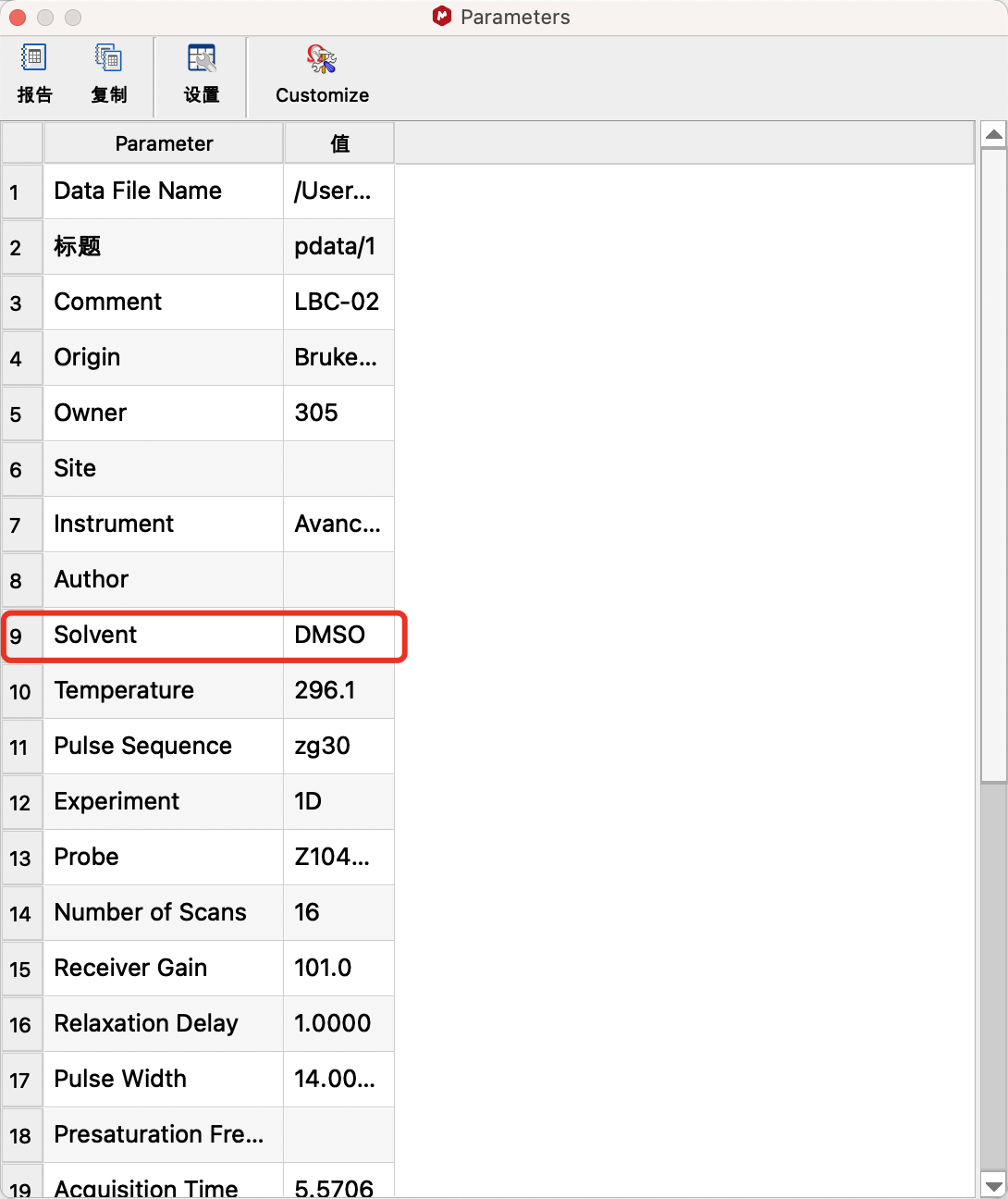
查看溶剂的标准化学位移:若化学位移0附近有明显的信号,可以使用TMS(化学位移等于0)进行定标。若其他溶剂的标准化学位移未知,可点击修正窗口中的Solvents(溶剂)查看对应的化学位移再进行定标。
具体操作:
1、放大需要定标的信号
2、点击Reference(参考)图标
3、将鼠标移动到对应信号上面(自动锁定顶点),点击左键,显示修正窗口
4、将New Shift修改为标准化学位移。
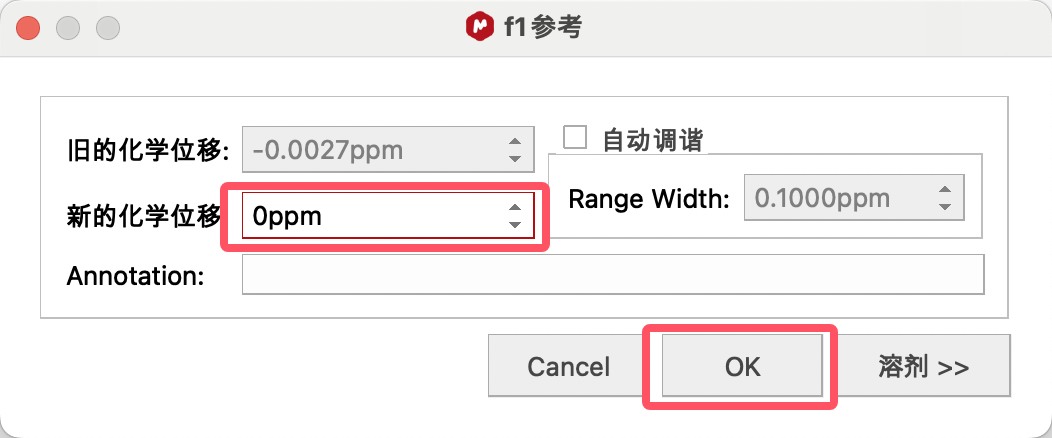
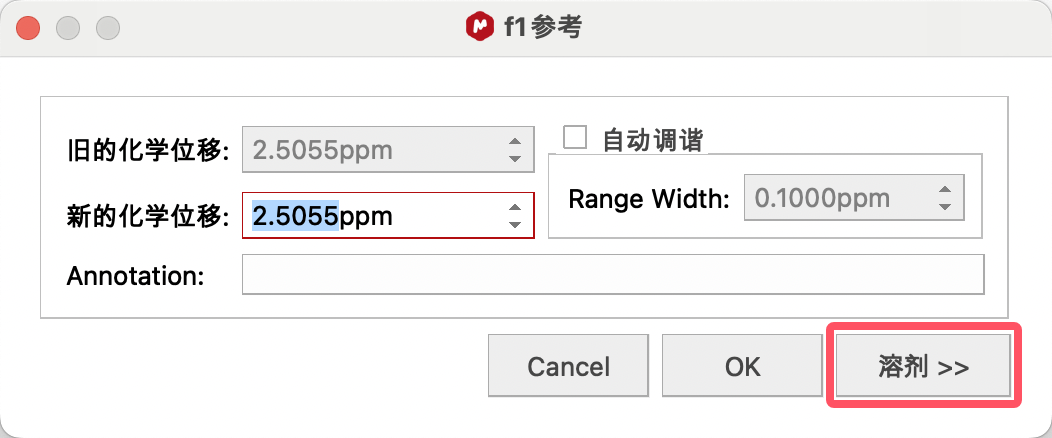
若溶剂峰的标准化学位移未知,可点击修正窗口中的Solvents(溶剂),查找溶剂的标准化学位移。
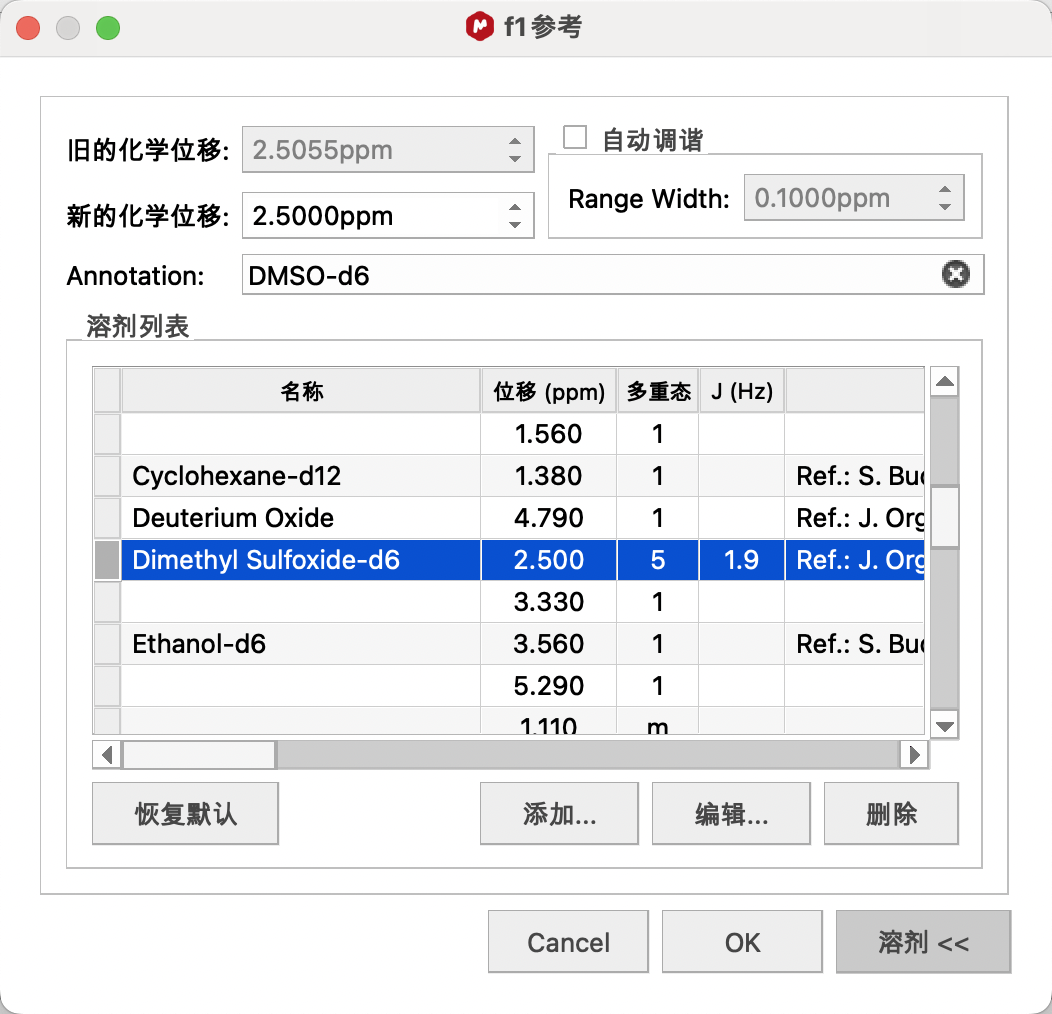
比如,要使用DMSO进行定标,选择DMSO的大致信号点(2.5ppm附近),点击修正窗口中的Solvents(溶剂),在其中找到DMSO的标准唯一,单击,点击OK进行定标。
(2)步骤二:Peaks(峰)——标记化学位移
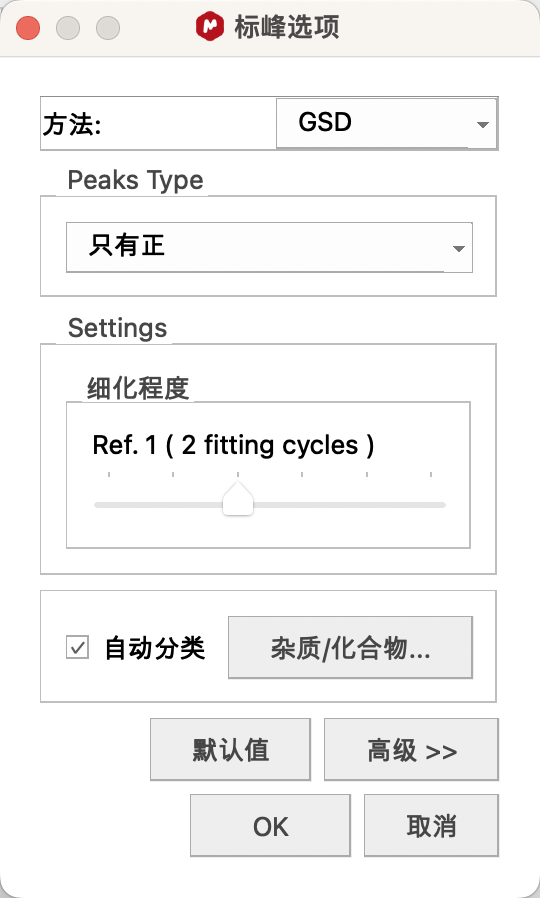
基础设置:点击Options(选项)图标,Method(方法)建议选择GSD(在标记化学位移时,软件还会对整张谱图进行全谱去卷积分析,信号中会出现蓝色线条,就是全谱去卷积拟合的结果,标记的化学位移值是以拟合的信号为准)。若不需要拟合出来的线,取消勾选Peak Curves。
勾选Auto Classify(自动分类),系统会自动区分化合物的信号和杂质的信号,将杂质的信号标红,同时在积分时剔除出去。
在具体标记化学位移时,有以下几种操作:
1、Auto Peak Picking——自动标记化学位移
点击Auto Peak Picking,系统会自动标记整张谱图的化学位移,其中红色是溶剂峰标识,但标记的数量太多,有很多没用的峰,不适用。
2、Manual Threshold——手动设置阈值
使用鼠标向上滚动,能更加容易清晰观察到有哪些是杂质比较低的信号,有哪些是需要的溶剂的信号。
点击Manual Threshold,把鼠标放在左侧,按着鼠标左键向右下角拖动,框选所有需要标记的信号(粉色区域),松开鼠标,软件会把处在粉色区域及以上的信号标记上化学位移,下方白色区域的小幅信号不标记。
使用右侧工具栏中的放大工具查看标峰情况。经过手动设置阈值标记后,若仍有部分信号未标记上,可以点击Pick by Pick(逐个标峰),一个个进行手动标记。若有不明显的峰不需要标记,可以点击Delete Manually(手动删除)删除不需要的化学位移。
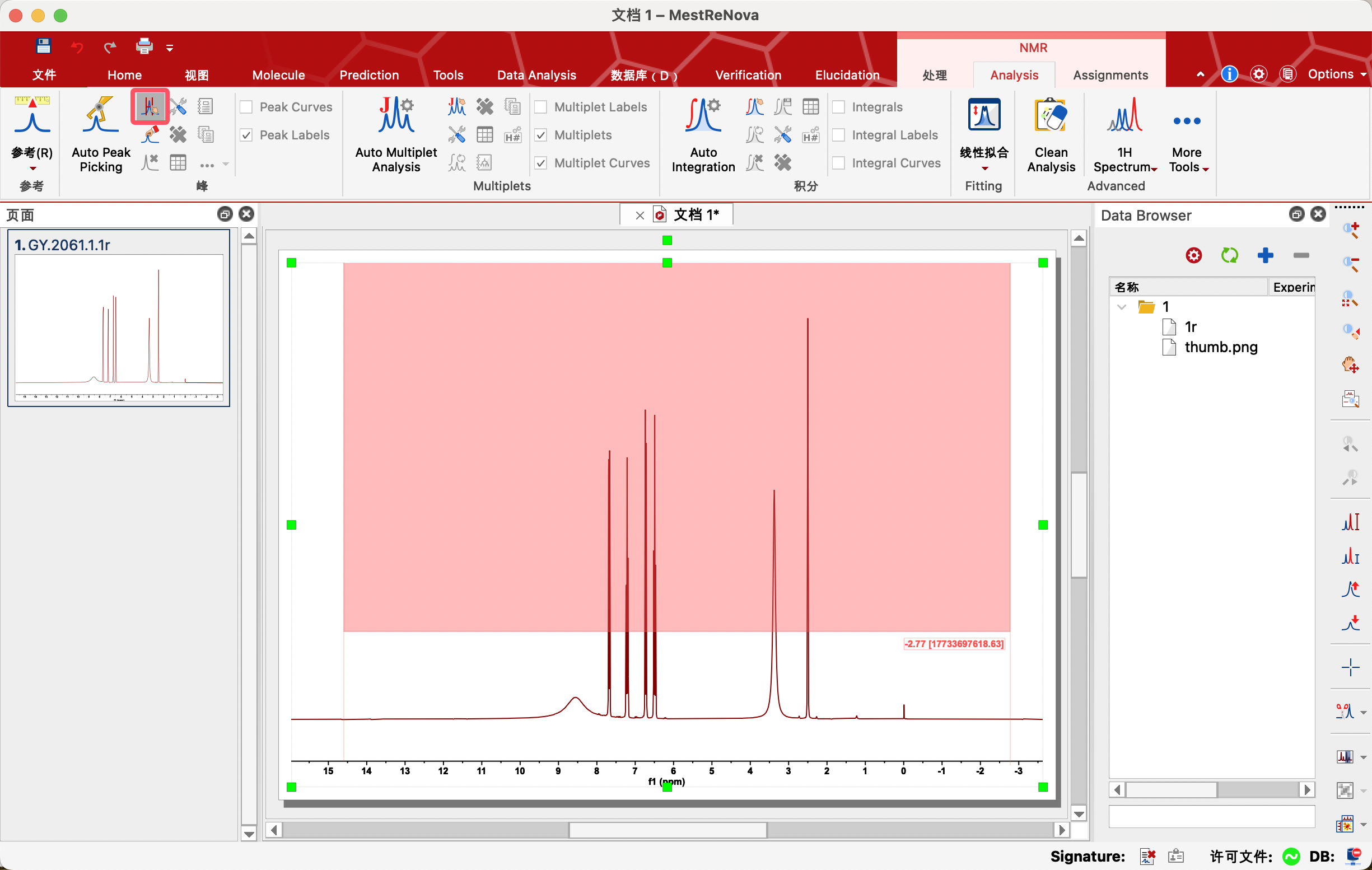
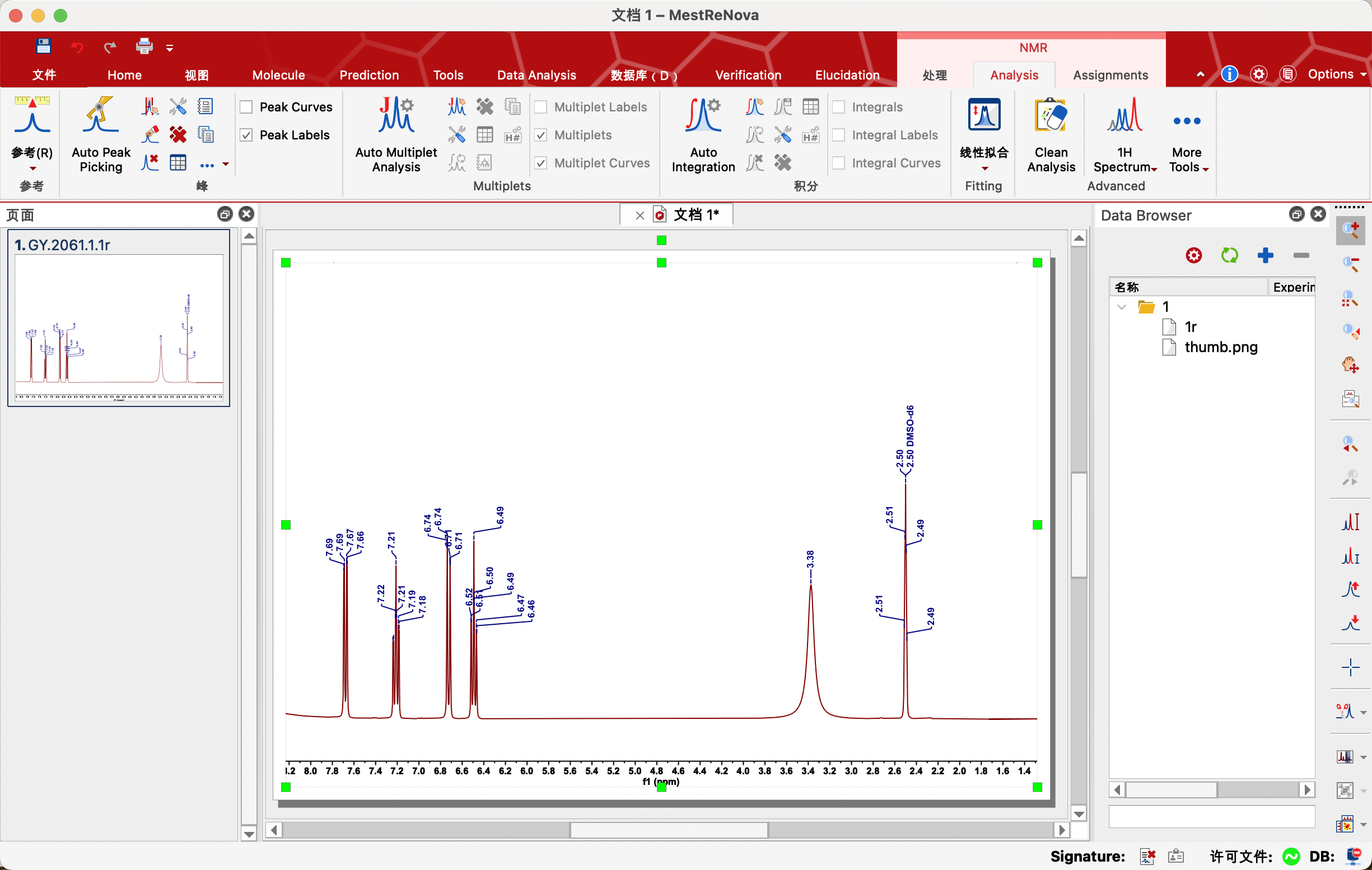
3、Peak by Peak——逐个标峰(建议使用)
由于我们所研究的物质是已知的,一般使用核磁氢谱来验证我们的产物。因此可以在网上查找所研究物质的核磁氢谱长啥样,与自己的谱图进行对比,手动标记出物质氢原子对应的峰。
核磁共振氢谱标准谱图查找方法:
1、使用chemdraw绘制化合物,点击菜单栏中Structure-Predict H-NMR Shifts进行核磁氢谱预测(不够准确)
2、在网上搜索化合物的核磁共振氢谱图,比如https://www.chemicalbook.com/。
3、通过scifinder等学术文献查找工具查找标准谱图。
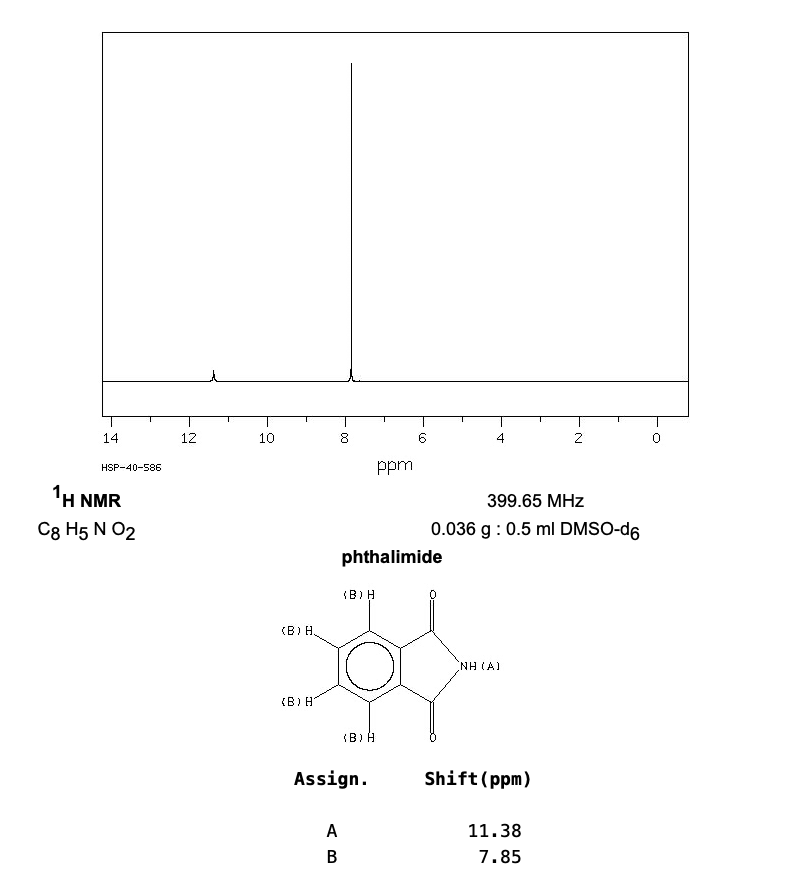
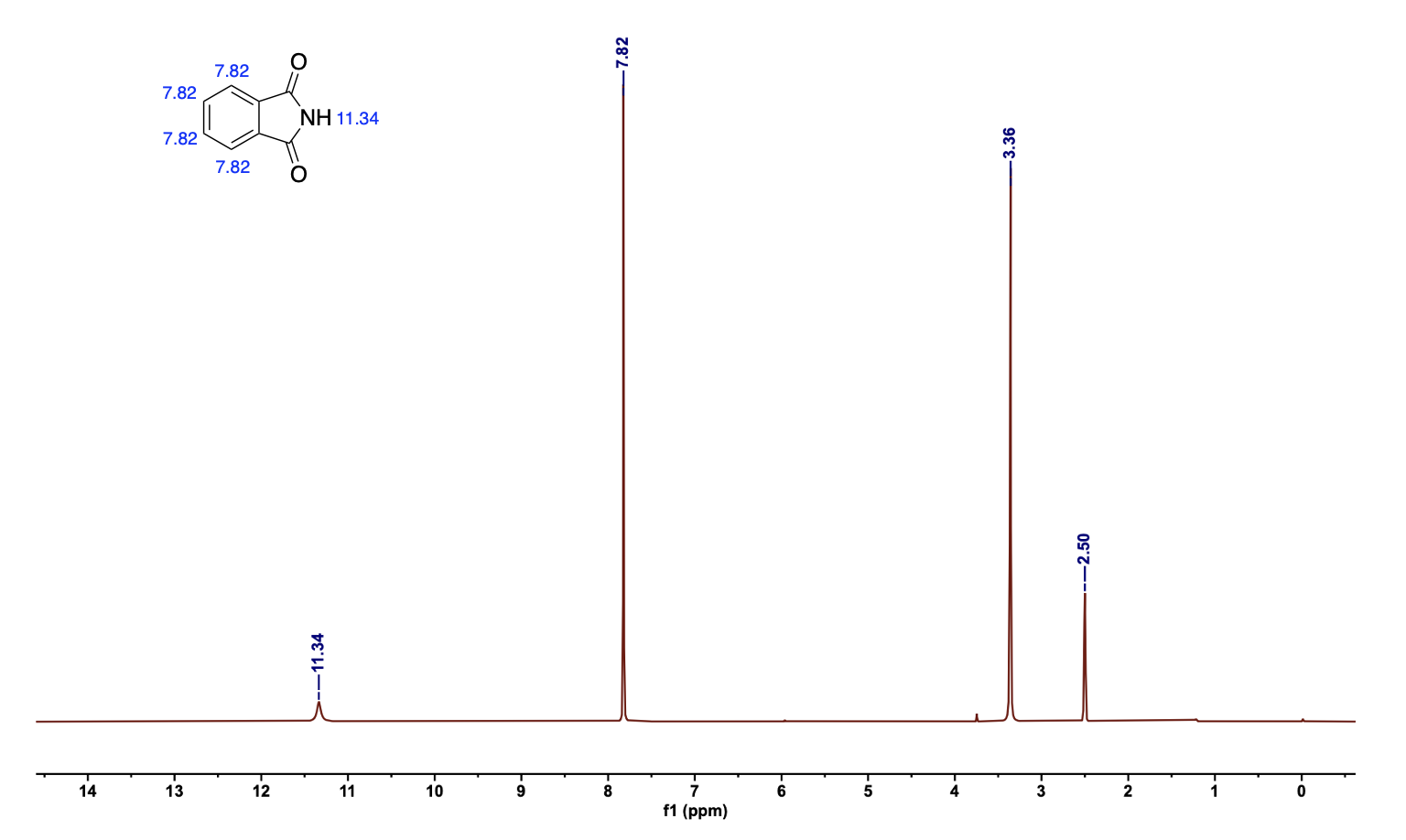
删除操作:
点击Delete All(全部删除),删除所以标记的化学位移。
点击Delete Manually(手动删除),手动删除不需要的化学位移。
其他操作:
点击NMR Peaks Table,显示一个表格,其中包含了所以标记的化学位移,并且对其进行归类,显示是杂质还是溶剂峰。
取消勾选Peak Curves,不显示GSD拟合的信号(蓝色线条)。
取消勾选Peak Labels,不显示标记的化学位移值。
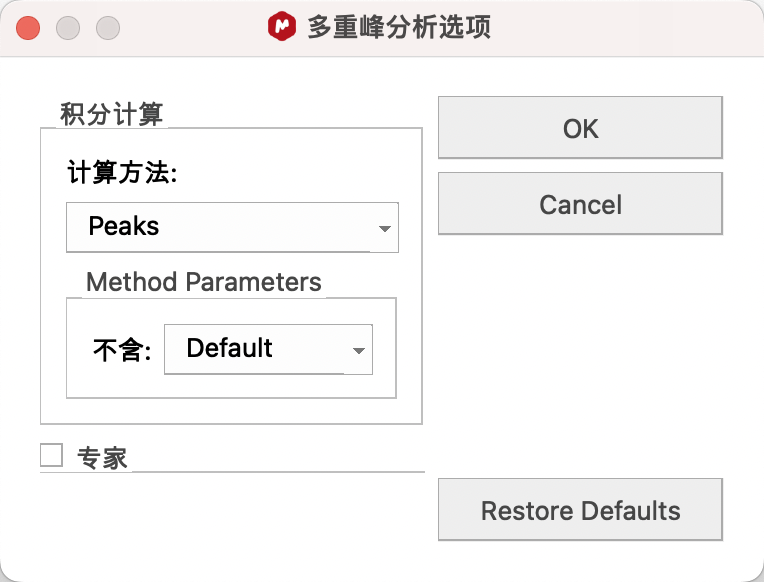
(3)步骤三:Multiplets(多重峰分析)或 Integration——积分
基础设置:点击Options,Calculation Method(积分计算方法)建议选择Peaks。
Sum积分方法计算信号线下的总面积(与GSD拟合结果无关),移动积分范围,积分值会改变。
Peaks积分方法将拟合出来的单峰面积相加(与GSD拟合结果有关),可以将信号进行拆分,移动积分范围,积分值不变。
1、Auto Multiplet Analysis——自动多重峰分析(建议使用)
多重峰分析不仅仅可以进行积分,自动做归一化,还可以判定峰的类型,标记最高峰的化学位移,并自动计算峰的耦合常数。
点击Auto Multiplet Analysis,自动对每一个信号进行多重峰分析(排除水和溶剂峰信号)。
归一化:将鼠标移到可判断出氢原子个数的信号处,双击该信号的多重峰分析结果(矩形框,积分值,积分线),弹出多重峰分析管理窗口,将积分改为已知的氢原子个数。
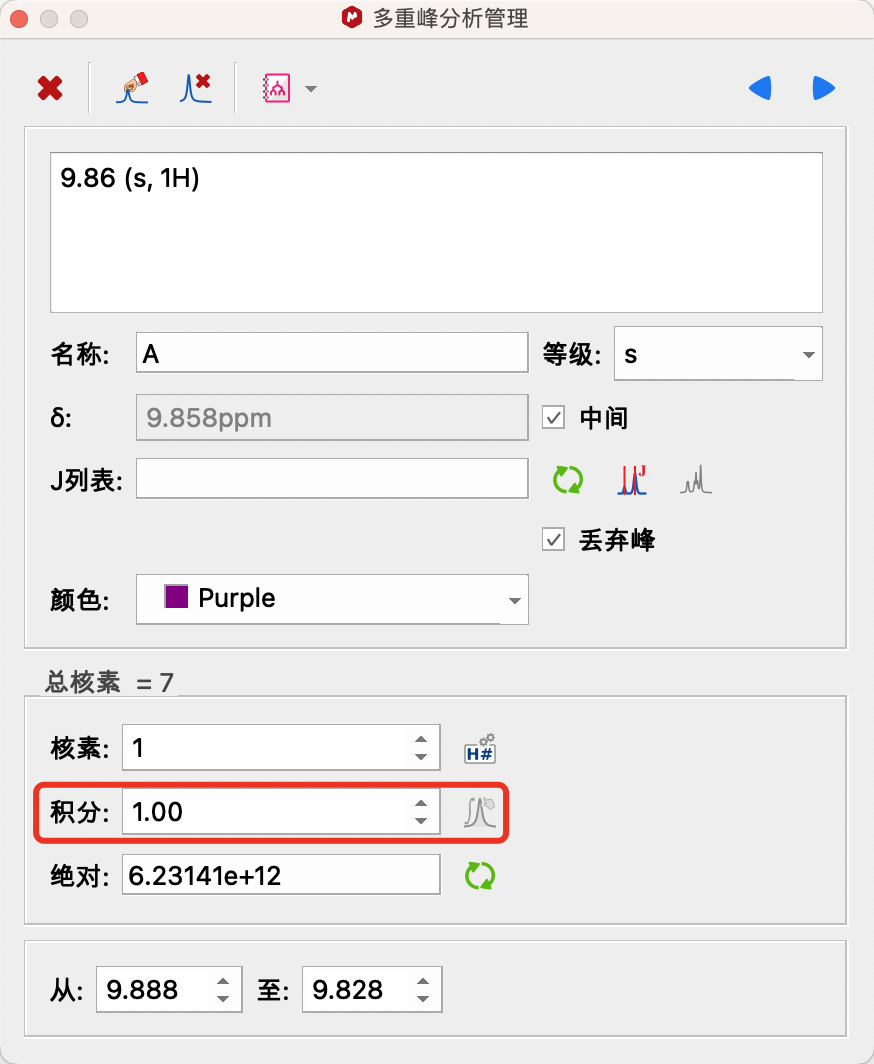
若需要对分析结果进行人为修改,可以双击信号的多重峰分析结果(矩形框,积分值,积分线)可对其进行修改。弹出的多重峰分析管理器会显示峰的所有信息,对于多重峰,存在耦合常数,点击Simulate Multiplets,软件可以根据得到的数据画出一个信号,与实际信号进行比较,用以检查。
点击Manually,对图谱进行手动积分。
点击NMR Multiplets Table,显示多重峰分析结果,可导出。
取消勾选Multiplet Labels,不显示多重峰分析得到的矩形框。
取消勾选Multiplets,不显示积分值。
取消勾选Multiplet Curves,不显示积分线。
2、Auto Integration——自动积分
仅能进行简单的积分,操作与标记化学位移类似。
三、图谱美化
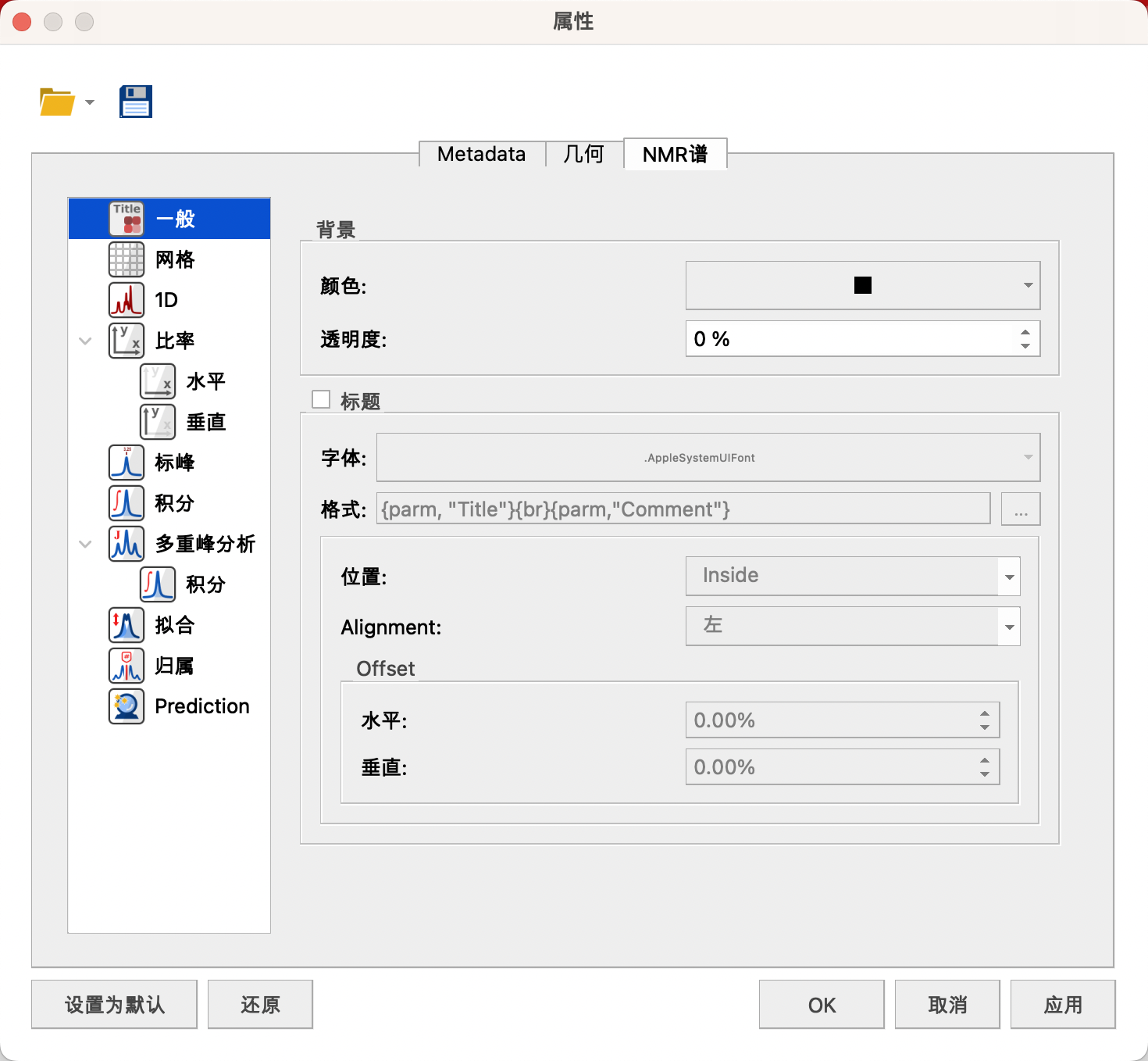
右键点击属性(Properties)选项,弹出属性窗口,
(1)Genera(一般)
背景:颜色——黑色
透明度——0%
取消勾选Title,不显示标题
Title中可设置字体,格式,位置,对齐方式和偏移
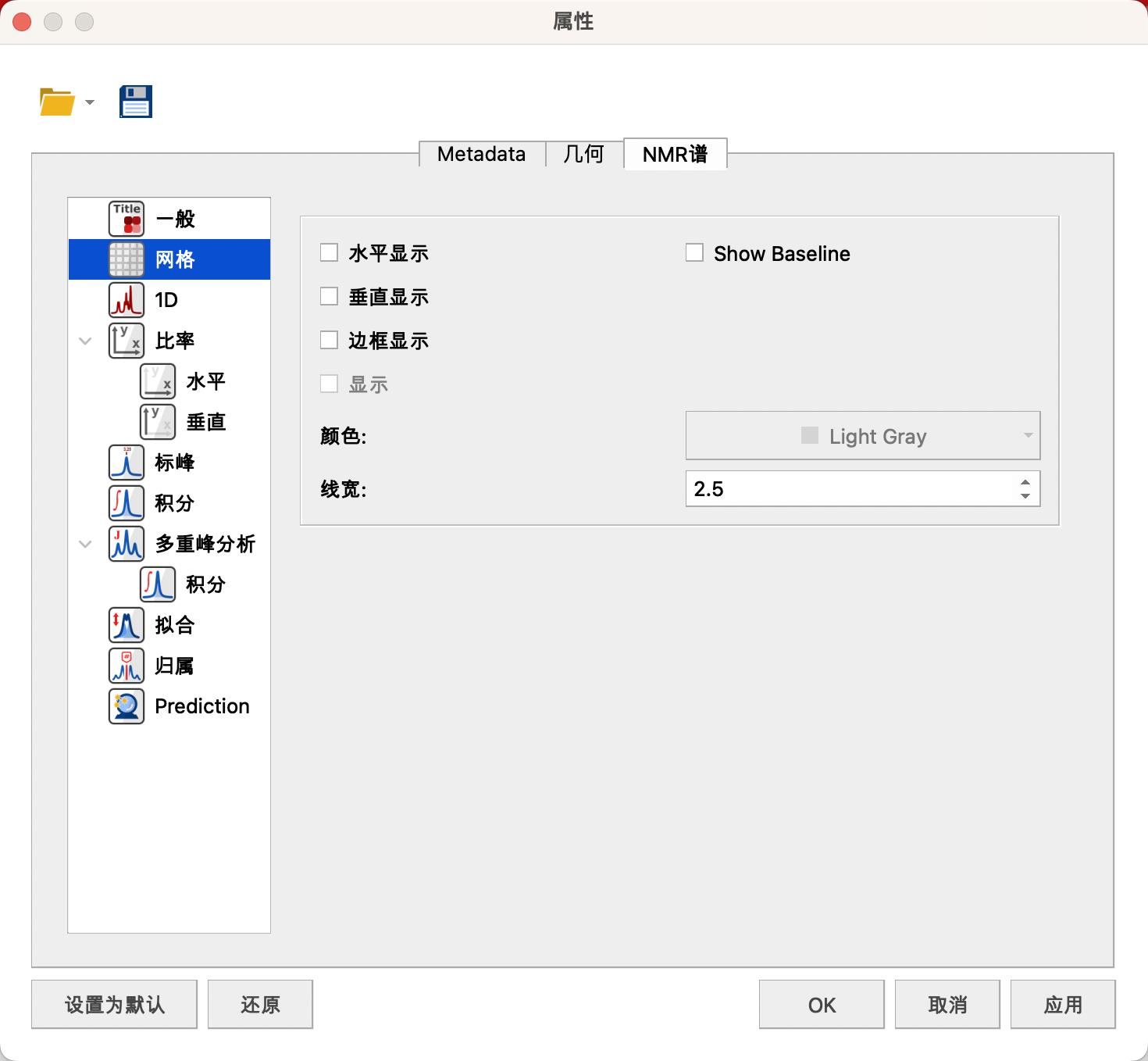
(2) Grid(网格)
取消勾选Show Horizontal,Show Vertical,不显示横纵表格。
取消勾选Show Frame,不显示边框
取消勾线Show Baseline,不显示基线
当勾选Show Over时,可以设置显示线条的颜色和宽度。
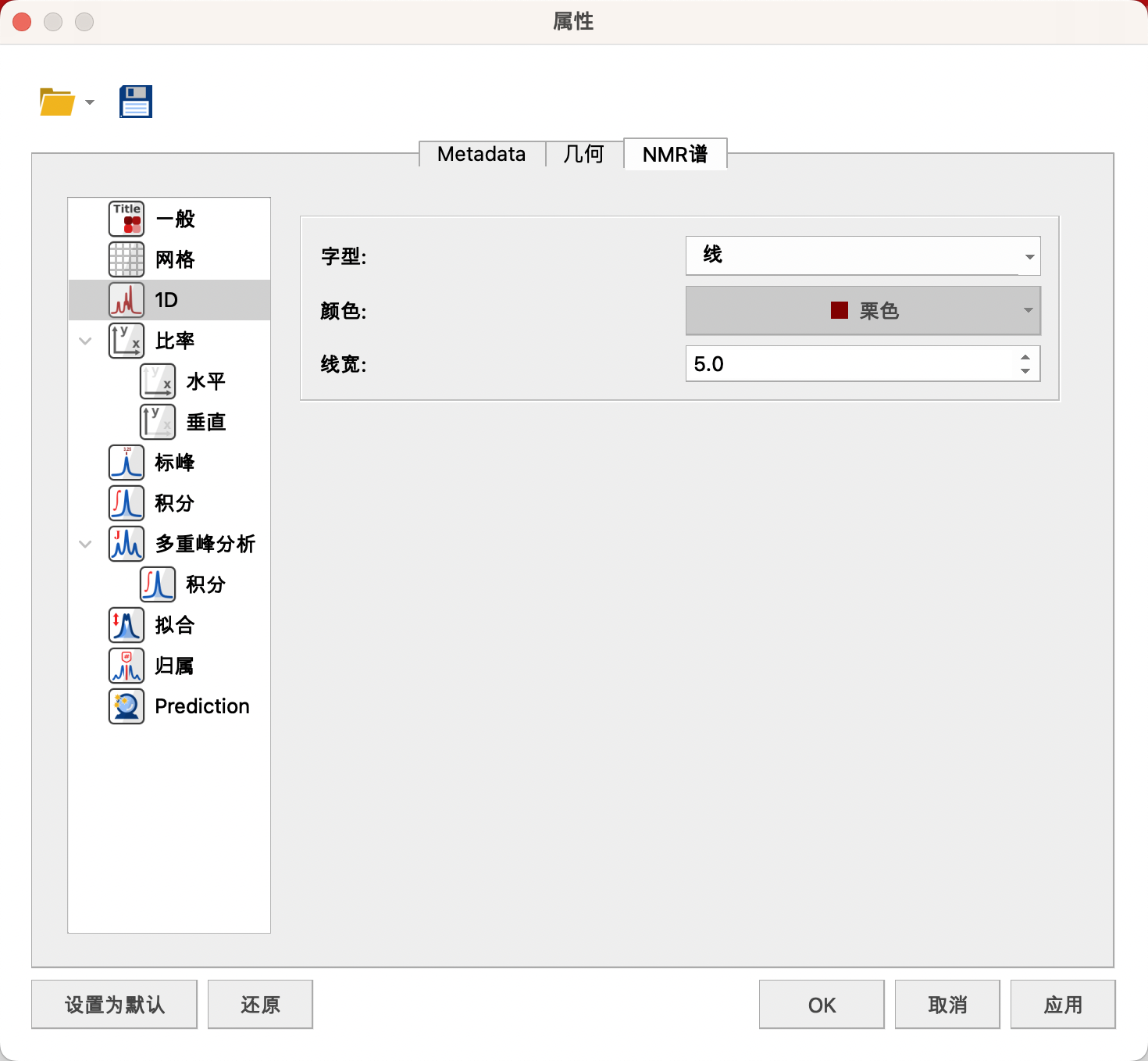
(3)1D(一维谱图显示)
设置谱线样式——线,颜色——栗色,线宽(电脑显示建议5,打印建议黑色1.0)
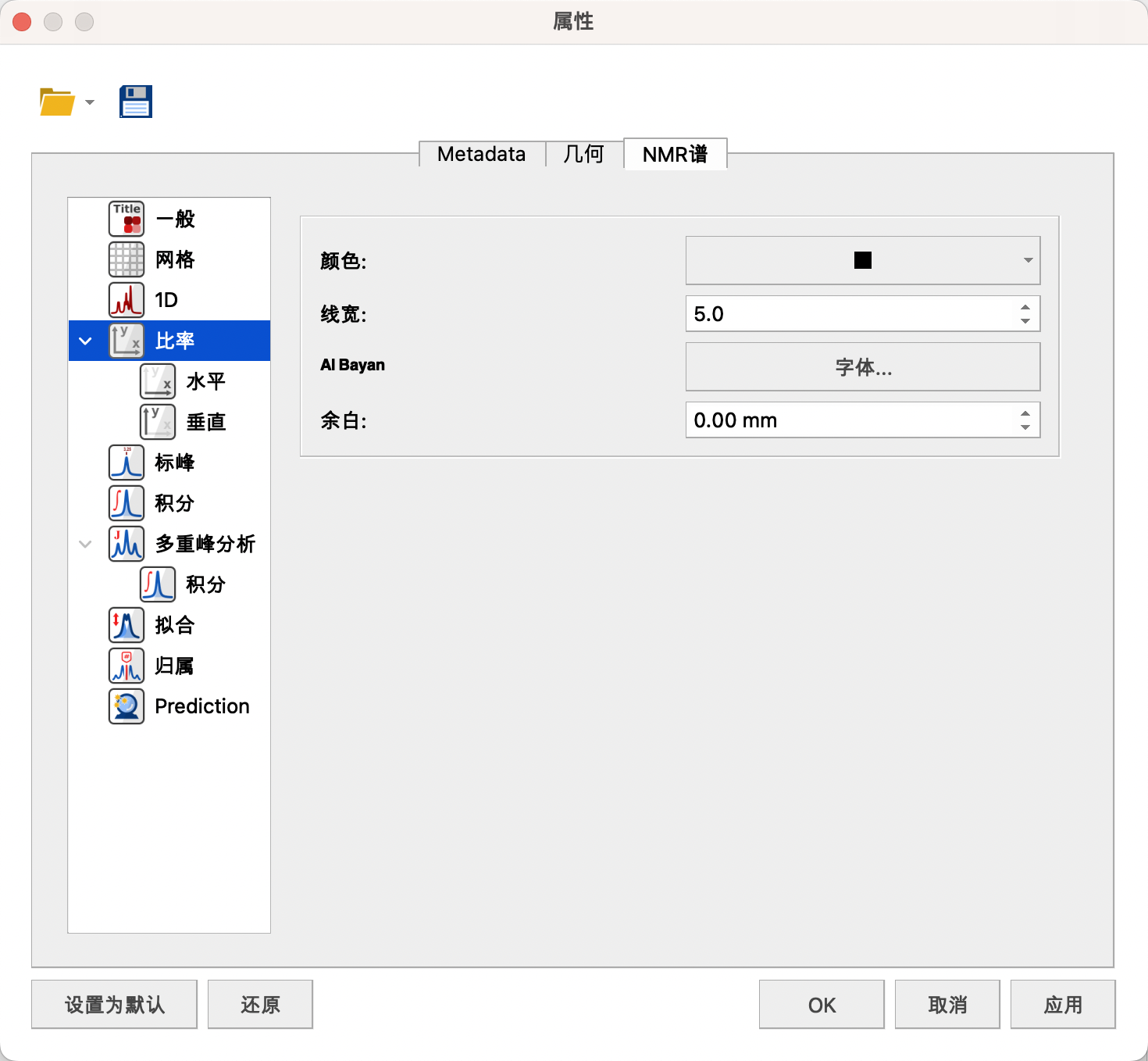
(4) Scales(比率)
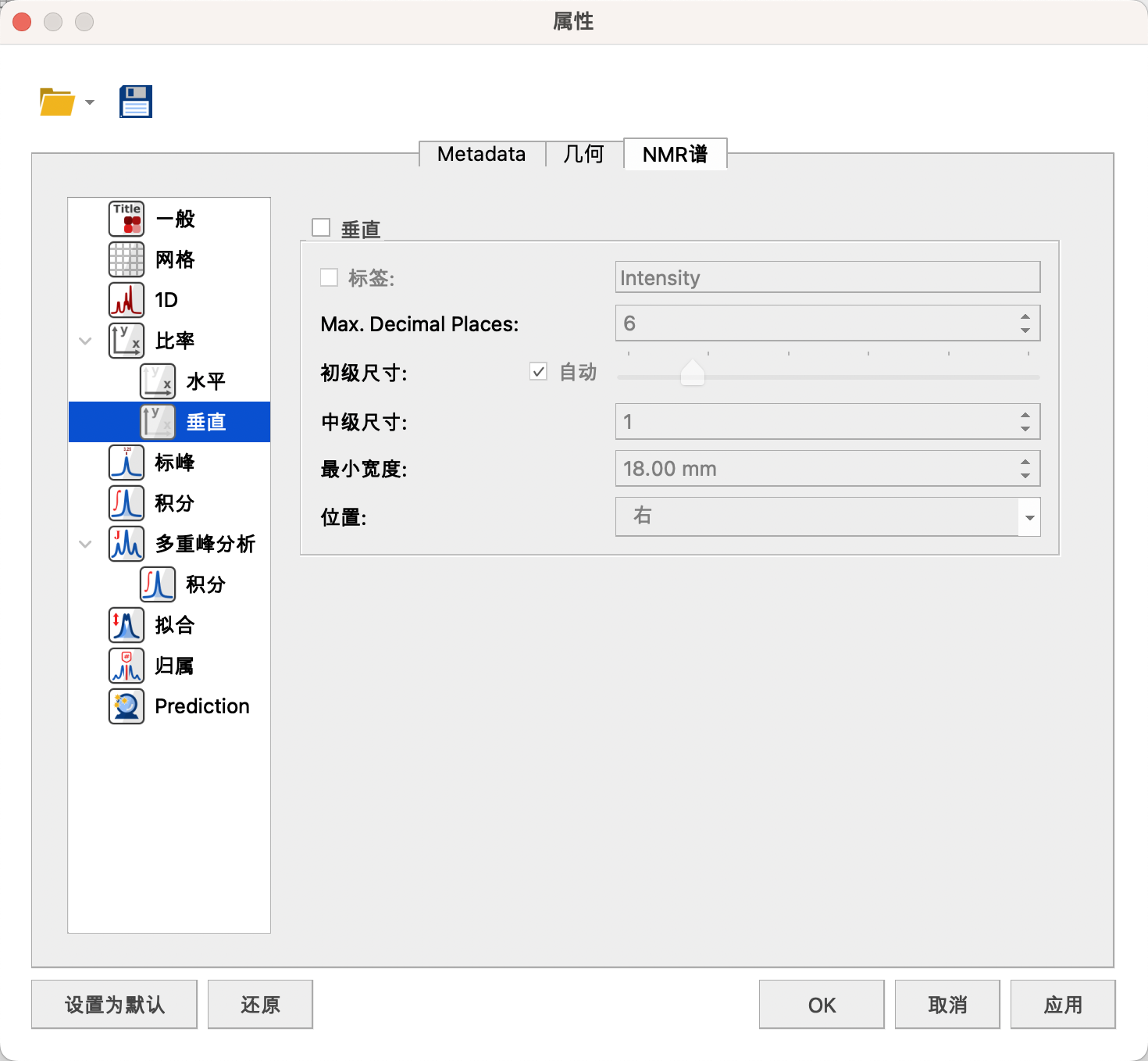
可以设置横纵坐标的颜色——黑色,线宽——5,字体——Arial或者Times New Roman,B,10号,谱图距离横坐标的距离——0。
一维谱图一般不显示纵坐标。
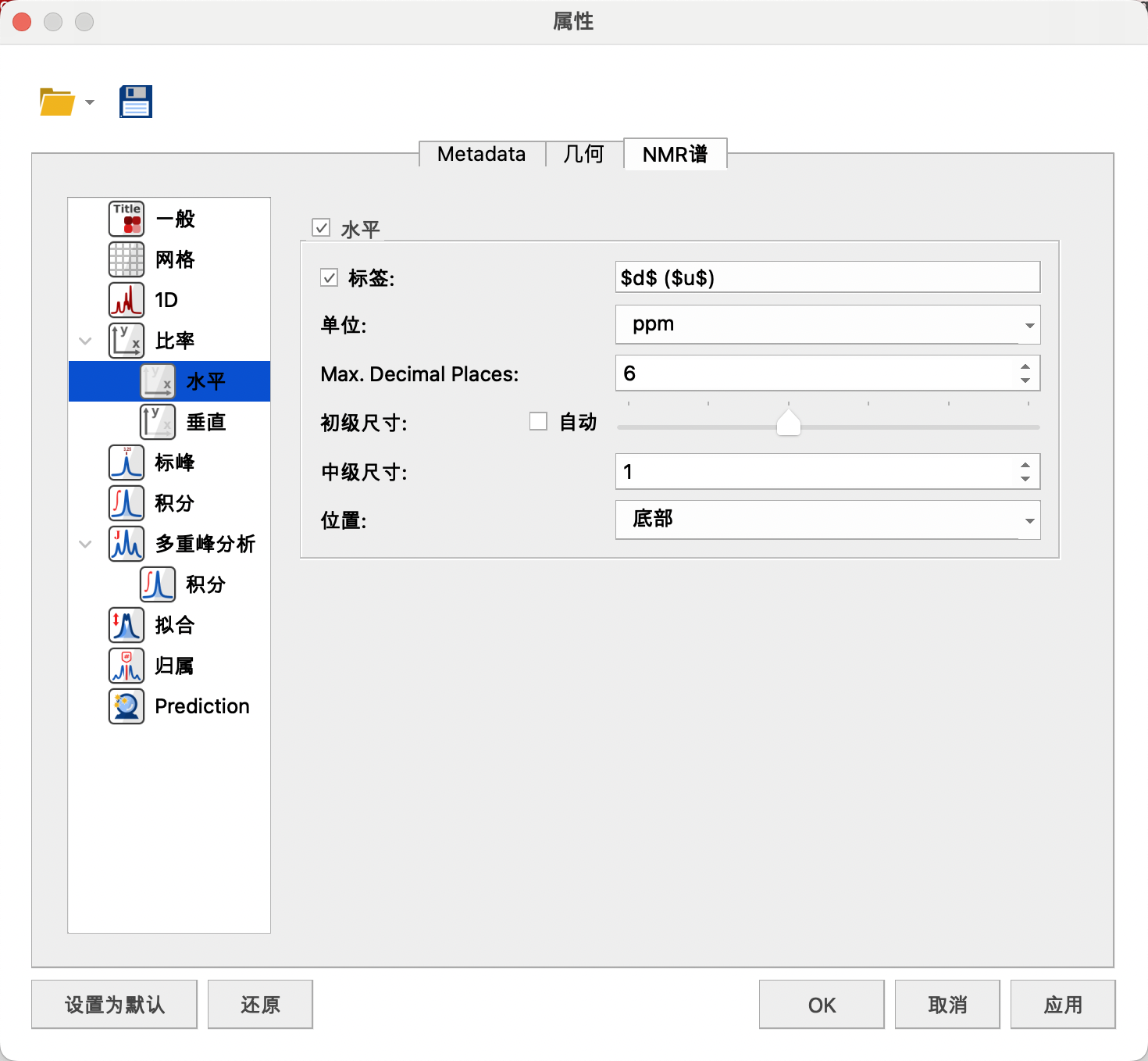
横纵坐标可以设置标签,单位——ppm,最大小数位——6,初级尺寸——s,中级尺寸——1,位置——底部。
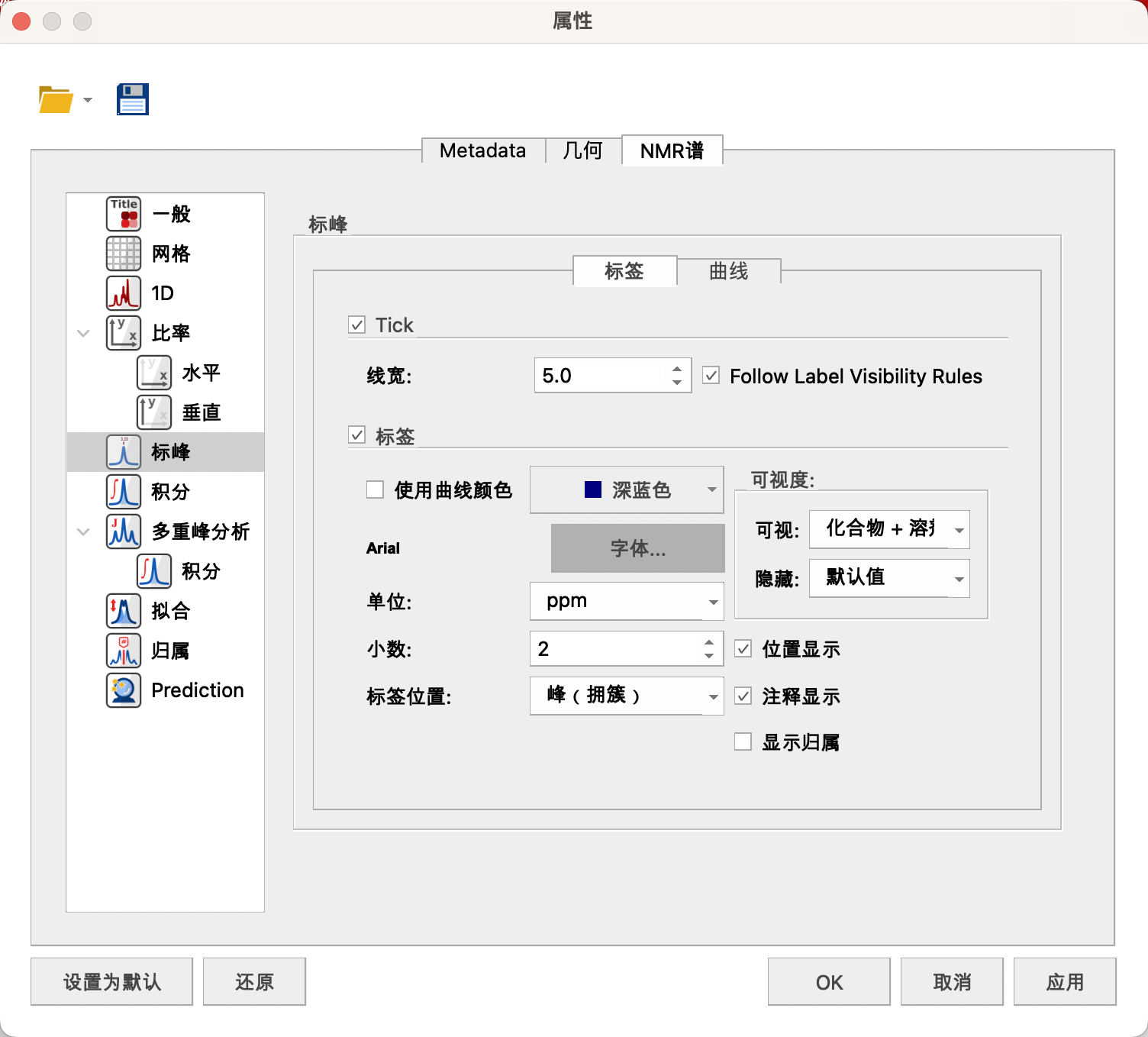
(5)Peaks(标峰)
可以设置线的宽度——5,颜色(取消勾选Use Curve Color)——深蓝色,字体的样式——Arial,B,10号,数字单位——ppm,小数——2,标记的位置——Peak Groups(峰(拥簇))
在Visibility中Visible的下拉菜单中勾选Compound和Solvent(仅显示化合物和溶剂的化学位移)
(6)Integrals(积分)
略
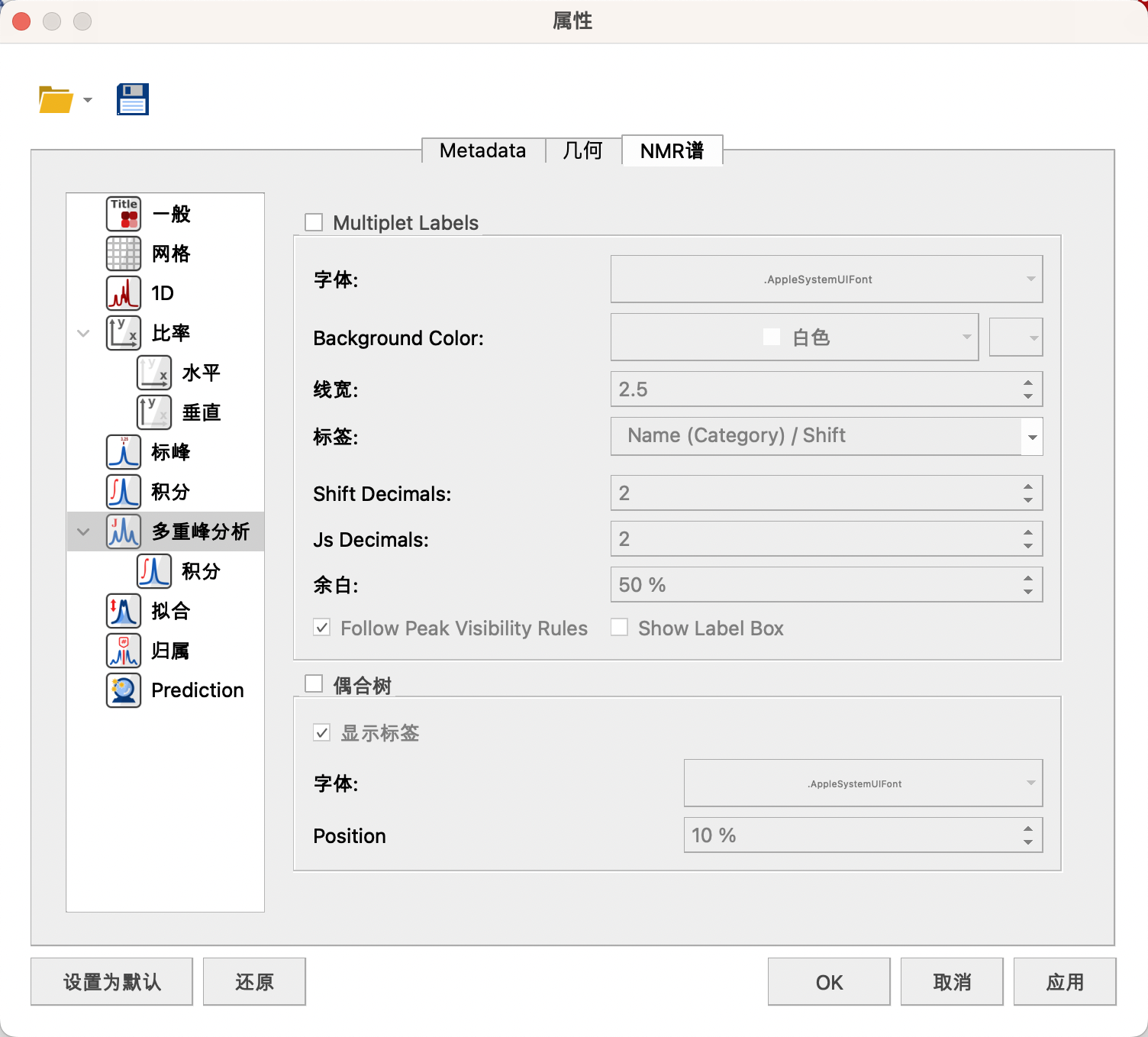
(7)Multiplets Integrals(多重峰分析)
可以设置矩形框字体样式,背景,线宽,标签,小数点位数,积分地方距离横坐标距离,(勾选J’s Tree(偶合树)会显示明显裂分信号的偶合方式)。
设置下方积分值处的颜色——深绿,线宽——5,字体——a,B,10号,navy深蓝色,小数位数——2,显示位置——线段上,显示方式——垂直,Range中的Margin处可设置积分值距离基线的距离——3%。
设置上分积线的位置——25%,最大高度——40%。
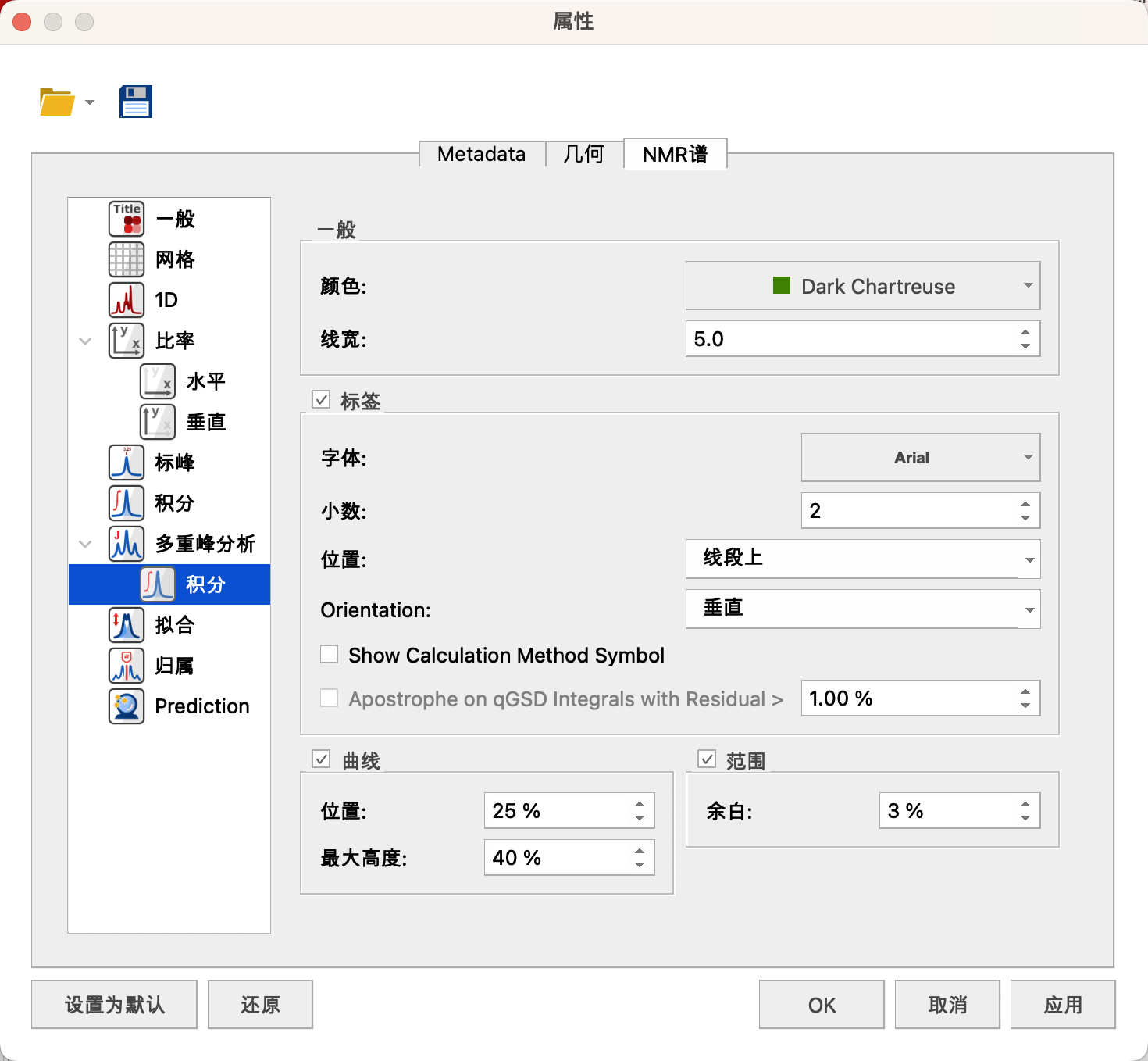
点击左下角Set as Default,设置成默认。
(8)谱线高度
用移动键将谱图基线移到适当高度了,积分数值位于基线下方。
滑动滚轮,将谱谱线高度移至页面一半以下。
用放大功能在包含所以峰的同时将谱图尽可能放大(0-10ppm)
导出为pdf文件。
四、调取相关信息
View中:
Tables中可以调取多重峰分析结果
勾选Parameters,显示谱图相关的原始信息(参数),可以在Customize自定义显示的信息
五、其他操作
Molecule处可以专门画结构,或用Chemdraw画图。
通过局部放大可以构建局部的另一副谱图
将整个页面作为一个模版可以点击View视图中的Layout Templates下的创建输出模版文档,同时可在此图标下粘贴模版。
第一次使用设置好基础选项后,对图谱分析基本流程:
导入图谱——自动基线校正——自动相位校正——查找溶剂型及其化学位移——定标——根据标准图谱逐个标峰进行定标——自动多重峰分析——归一化——图形美化——导出。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号