

Post-GWAS: single-cell disease relevance score (scDRS) 分析
1、scDRS的计算原理如下所示:
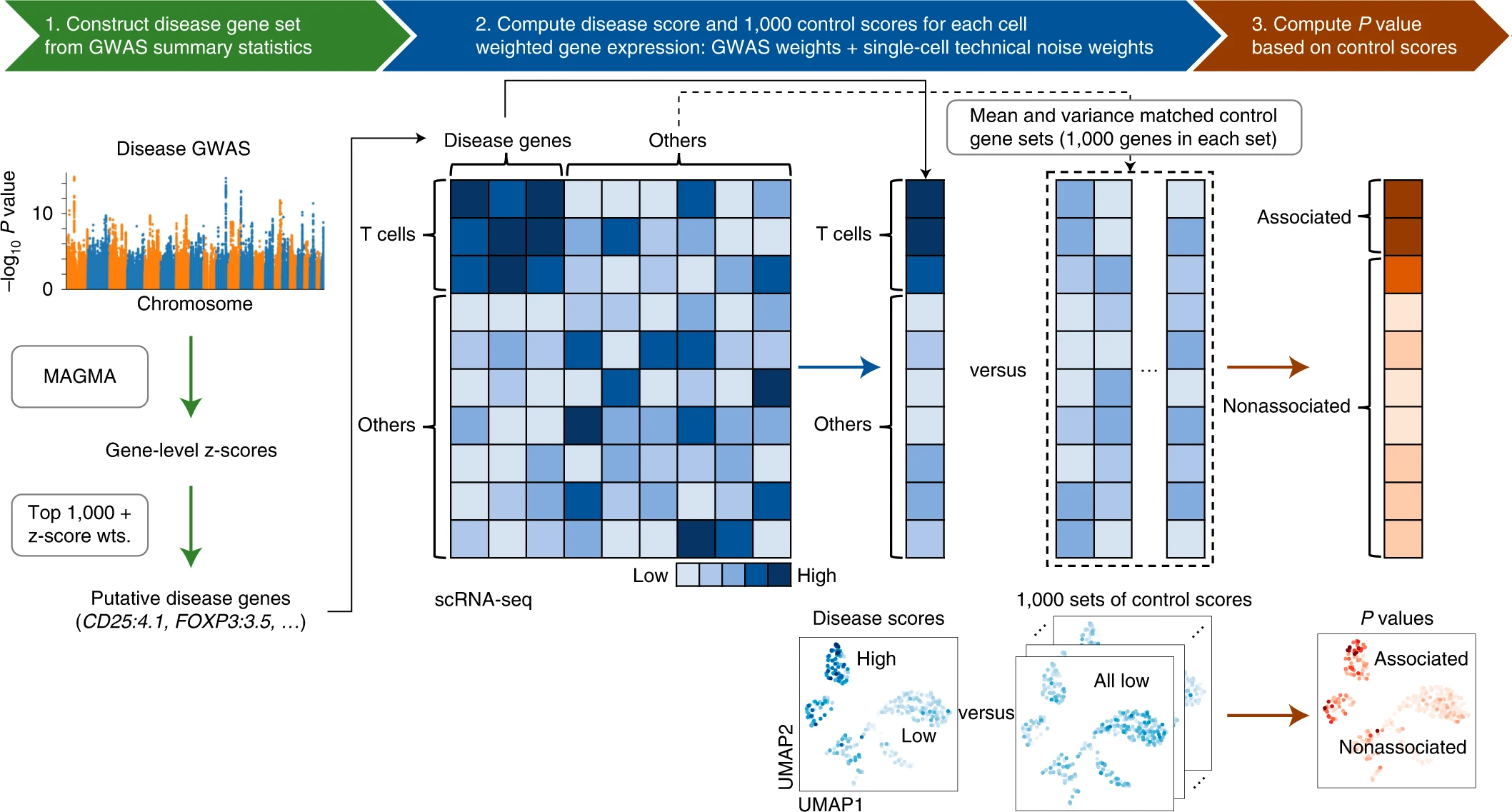
图片来源:Zhang M J, Hou K, Dey K K, et al. Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data[R]. Nature Publishing Group, 2022.
2、通过scDRS分析可以得到什么
第一、鉴定表型相关的细胞类型,比如下图:
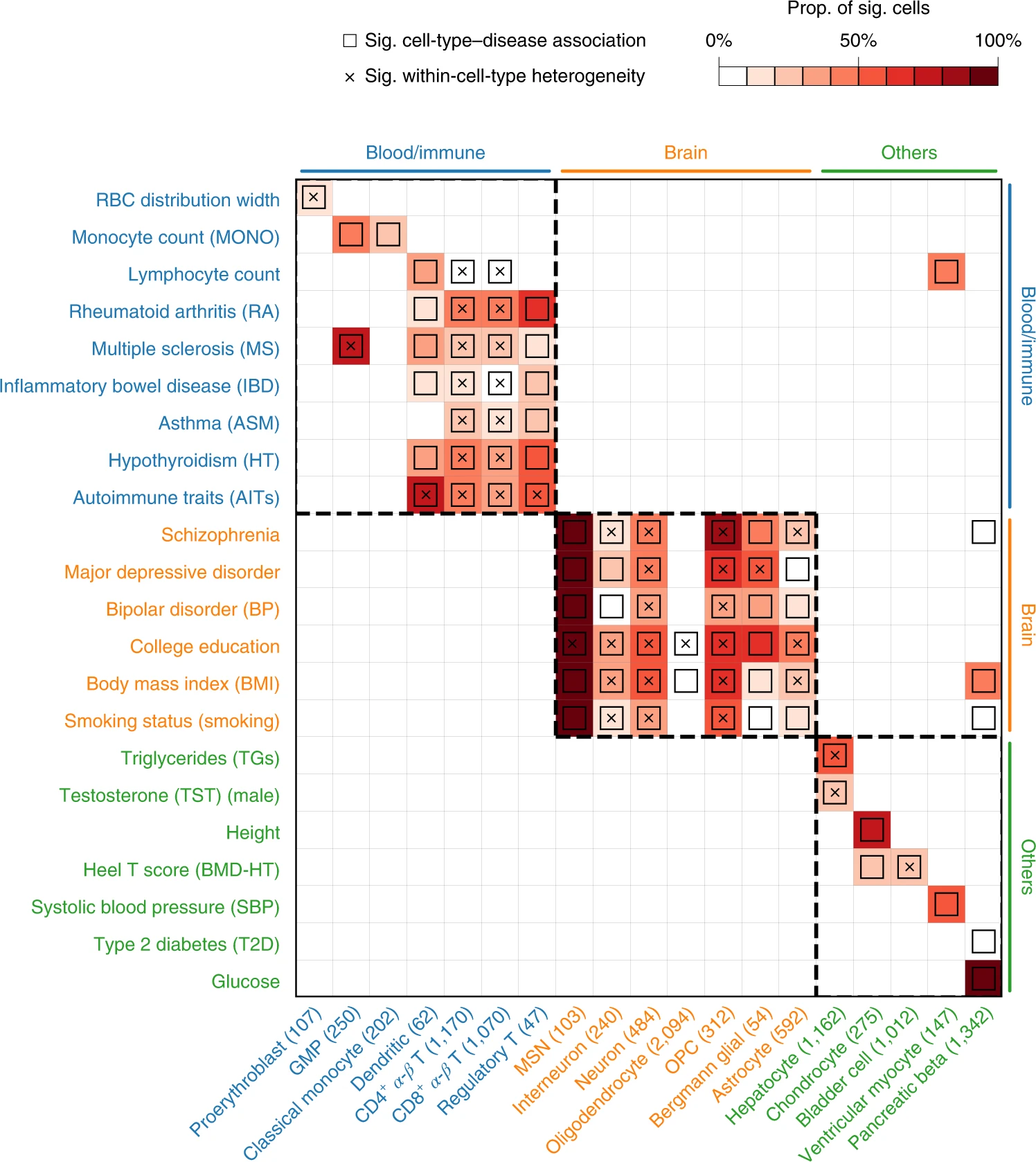
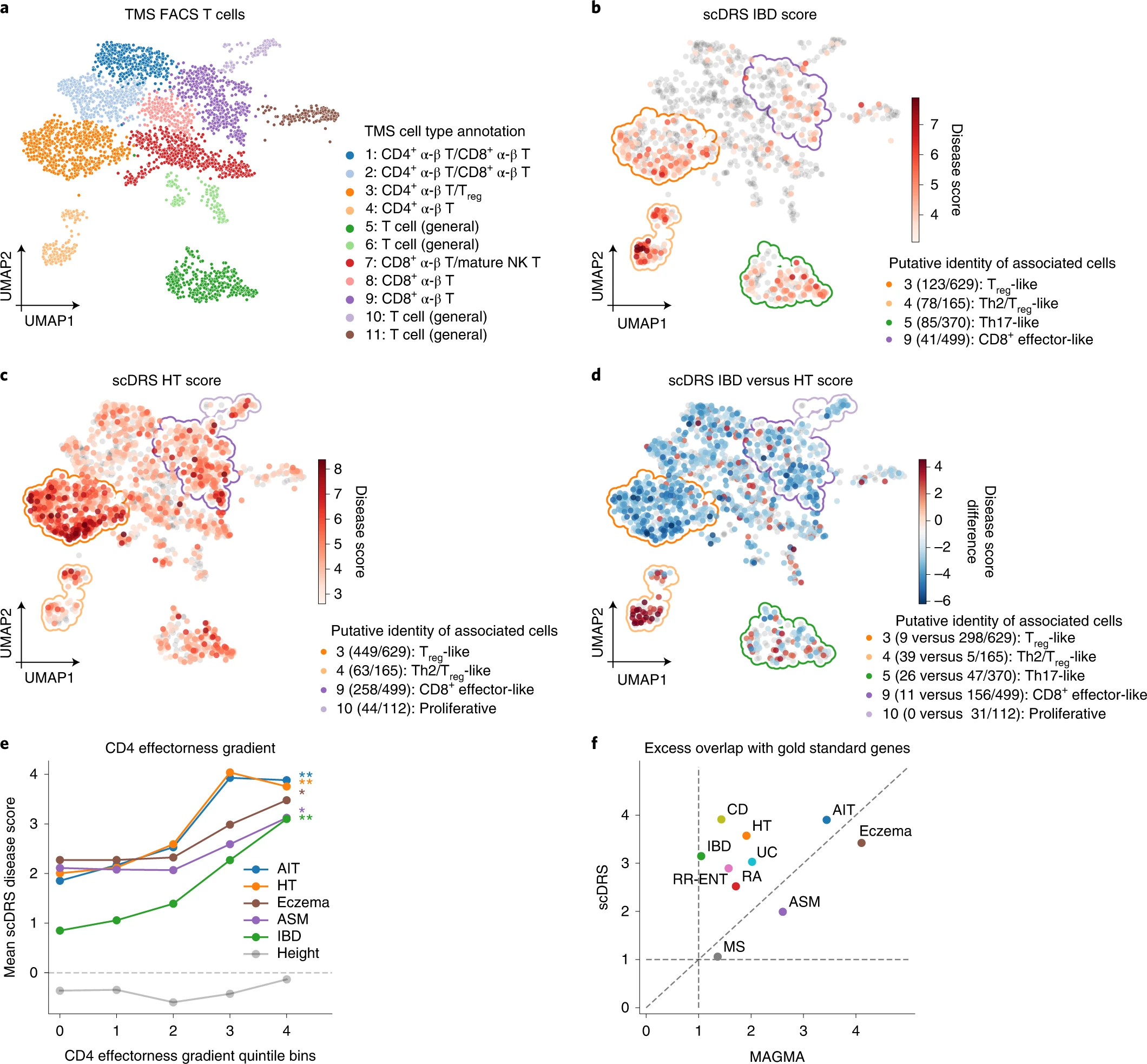
第二、发现疾病的subpopulations:
3、scDRS安装与运行
3.1 下载、安装
#下载安装jupyter
pip3 install jupyter
#下载安装scDRS
git clone https://github.com/martinjzhang/scDRS.git
cd scDRS
git checkout -b v102 v1.0.2
pip install -e .
3.2 测试安装成功了没有
python -m pytest tests/test_CLI.py -p no:warnings
3.3 下载测试数据
wget https://figshare.com/ndownloader/files/34300925 -O data.zip
unzip data.zip && \
mkdir -p data/ && \
mv single_cell_data/zeisel_2015/* data/ && \
rm data.zip && rm -r single_cell_data
3.4 加载环境
#打开jupyter
jupyter notebook
打开jupyter后,我们转战到jupyter中工作。注意,后续所有的分析均在jupyter中实现。
#在jupyter中输入
import scdrs
import scanpy as sc
sc.set_figure_params(dpi=125)
from anndata import AnnData
from scipy import stats
import pandas as pd
import numpy as np
import seaborn as sns
import matplotlib.pyplot as plt
import os
import warnings
warnings.filterwarnings("ignore")
3.5 使用 MAGMA 计算疾病在基因水平的关联水平
该步骤参考教程:# 基于 MAGMA 的 gene-based 关联分析研究
得到gene-based 水平的zscore值后,将其整理成如下格式,文件名为geneset.zscore:
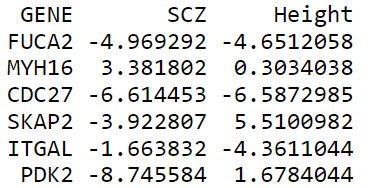
第一列是基因名,第二列是SCZ的gene-based zscore值,第三列是Height的gene-based zscore值;
3.6 将 MAGMA 输出的结果转为scDRS的格式
# Select top 1,000 genes and use z-score weights
!scdrs munge-gs \
--out-file single_cell_data/zeisel_2015/processed_geneset.gs \
--zscore-file single_cell_data/zeisel_2015/geneset.zscore \
--weight zscore \
--n-max 1000
结果文件processed_geneset.gs如下所示:

3.7 计算每个细胞的表型富集度
%%capture
#新建一个输出文件夹
!mkdir -p single_cell_data/cwy
#开始计算富集度
!scdrs compute-score \
--h5ad-file single_cell_data/zeisel_2015/expr.h5ad \
--h5ad-species mouse \
--gs-file single_cell_data/zeisel_2015/processed_geneset.gs \
--gs-species mouse \
--cov-file data/cov.tsv \
--flag-filter-data True \
--flag-raw-count True \
--flag-return-ctrl-raw-score False \
--flag-return-ctrl-norm-score True \
--out-folder single_cell_data/cwy/
3.8 可视化scDRS结果
dict_score = {
trait: pd.read_csv(f"single_cell_data/cwy/{trait}.full_score.gz", sep="\t", index_col=0)
for trait in df_gs.index
}
for trait in dict_score:
adata.obs[trait] = dict_score[trait]["norm_score"]
sc.set_figure_params(figsize=[2.5, 2.5], dpi=150)
sc.pl.umap(
adata,
color="level1class",
ncols=1,
color_map="RdBu_r",
vmin=-5,
vmax=5,
)
sc.pl.umap(
adata,
color=dict_score.keys(),
color_map="RdBu_r",
vmin=-5,
vmax=5,
s=20,
)
结果如下所示:
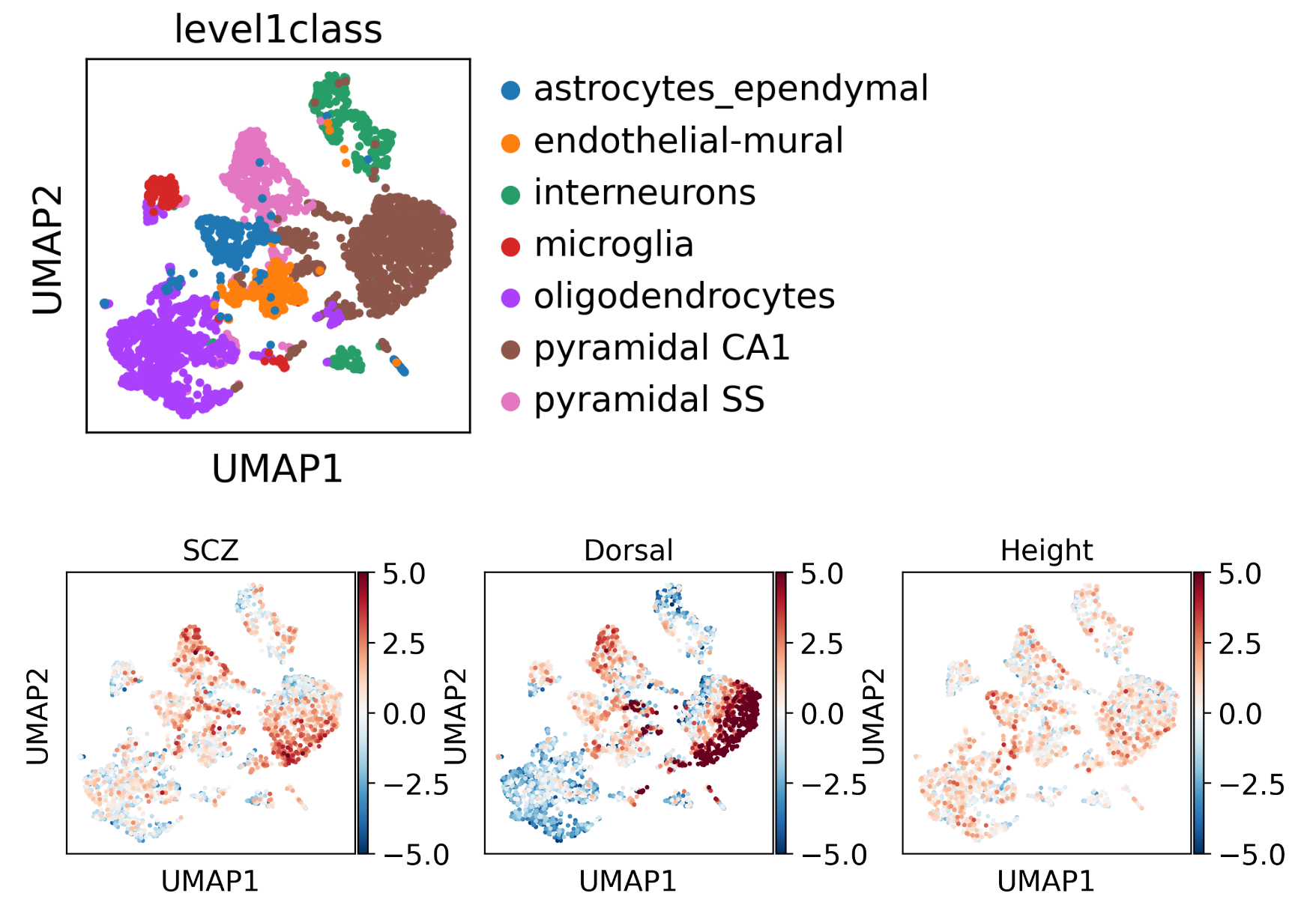
可见,SCZ疾病注意富集在Pyramidal CA1细胞;
3.9 分析疾病在不同细胞的表达水平差异
for trait in ["SCZ", "Height"]:
!scdrs perform-downstream \
--h5ad-file single_cell_data/zeisel_2015/expr.h5ad \
--score-file single_cell_data/cwy/{trait}.full_score.gz \
--out-folder single_cell_data/cwy/ \
--group-analysis level1class \
--flag-filter-data True \
--flag-raw-count True
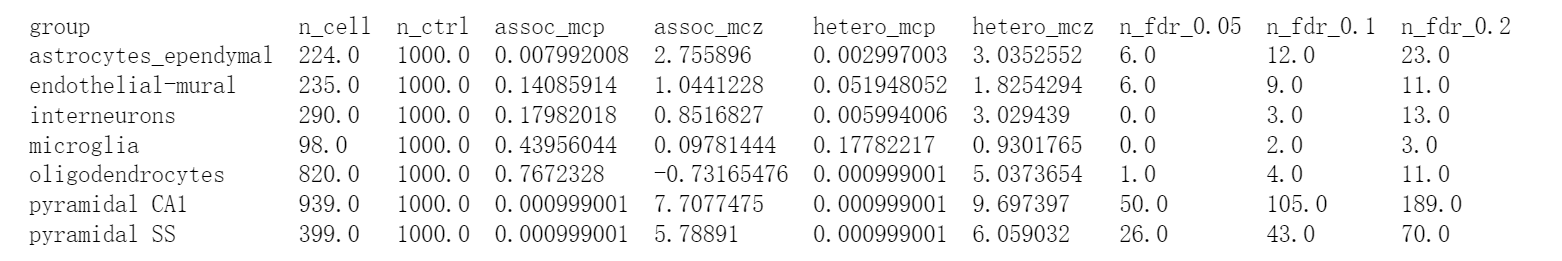
SCZ在不同细胞的表达水平差异:
# scDRS group-level statistics for SCZ
!cat single_cell_data/cwy/SCZ.scdrs_group.level1class | column -t -s $'\t'
可以看到,SCZ在Pyramidal CA1细胞表达较多;
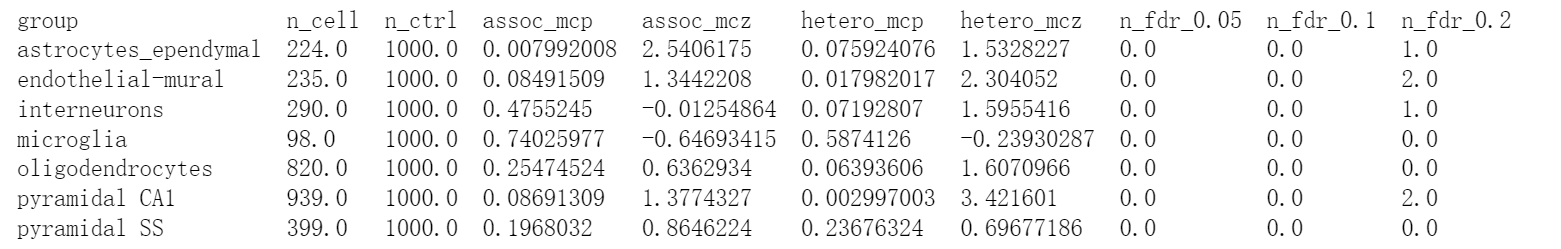
Height在不同细胞的表达水平差异:
# scDRS group-level statistics for Height
!cat single_cell_data/cwy/Height.scdrs_group.level1class | column -t -s $'\t'
3.10 对疾病在不同细胞的表达水平差异进行可视化
dict_df_stats = {
trait: pd.read_csv(f"single_cell_data/cwy/{trait}.scdrs_group.level1class", sep="\t", index_col=0)
for trait in ["SCZ", "Height"]
}
dict_celltype_display_name = {
"pyramidal_CA1": "Pyramidal CA1",
"oligodendrocytes": "Oligodendrocyte",
"pyramidal_SS": "Pyramidal SS",
"interneurons": "Interneuron",
"endothelial-mural": "Endothelial",
"astrocytes_ependymal": "Astrocyte",
"microglia": "Microglia",
}
fig, ax = scdrs.util.plot_group_stats(
dict_df_stats={
trait: df_stats.rename(index=dict_celltype_display_name)
for trait, df_stats in dict_df_stats.items()
},
plot_kws={
"vmax": 0.2,
"cb_fraction":0.12
}
)
可视化结果如下所示:
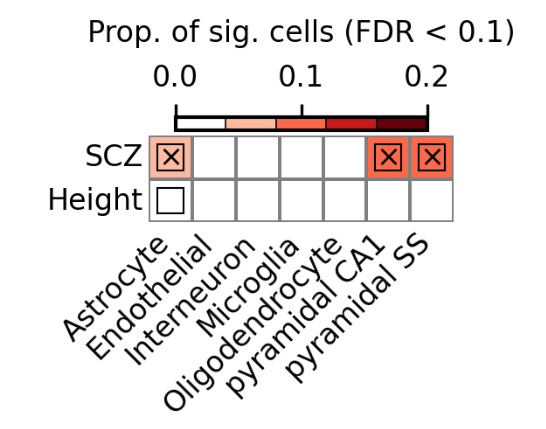
可以看到CA1 pyramidal在SCZ和height的差异是最明显的。
因此接下来就提取CA1 pyramidal细胞进行重聚类,解析导致SCZ和height差异的细胞。
3.11 解析不同疾病在某类细胞的异质来源
# extract CA1 pyramidal neurons and perform a re-clustering
adata_ca1 = adata[adata.obs["level2class"].isin(["CA1Pyr1", "CA1Pyr2"])].copy()
sc.pp.filter_cells(adata_ca1, min_genes=0)
sc.pp.filter_genes(adata_ca1, min_cells=1)
sc.pp.normalize_total(adata_ca1, target_sum=1e4)
sc.pp.log1p(adata_ca1)
sc.pp.highly_variable_genes(adata_ca1, min_mean=0.0125, max_mean=3, min_disp=0.5)
adata_ca1 = adata_ca1[:, adata_ca1.var.highly_variable]
sc.pp.scale(adata_ca1, max_value=10)
sc.tl.pca(adata_ca1, svd_solver="arpack")
sc.pp.neighbors(adata_ca1, n_neighbors=10, n_pcs=40)
sc.tl.umap(adata_ca1, n_components=2)
# assign scDRS score
for trait in dict_score:
adata_ca1.obs[trait] = dict_score[trait]["norm_score"]
sc.pl.umap(
adata_ca1,
color=dict_score.keys(),
color_map="RdBu_r",
vmin=-5,
vmax=5,
s=20,
)
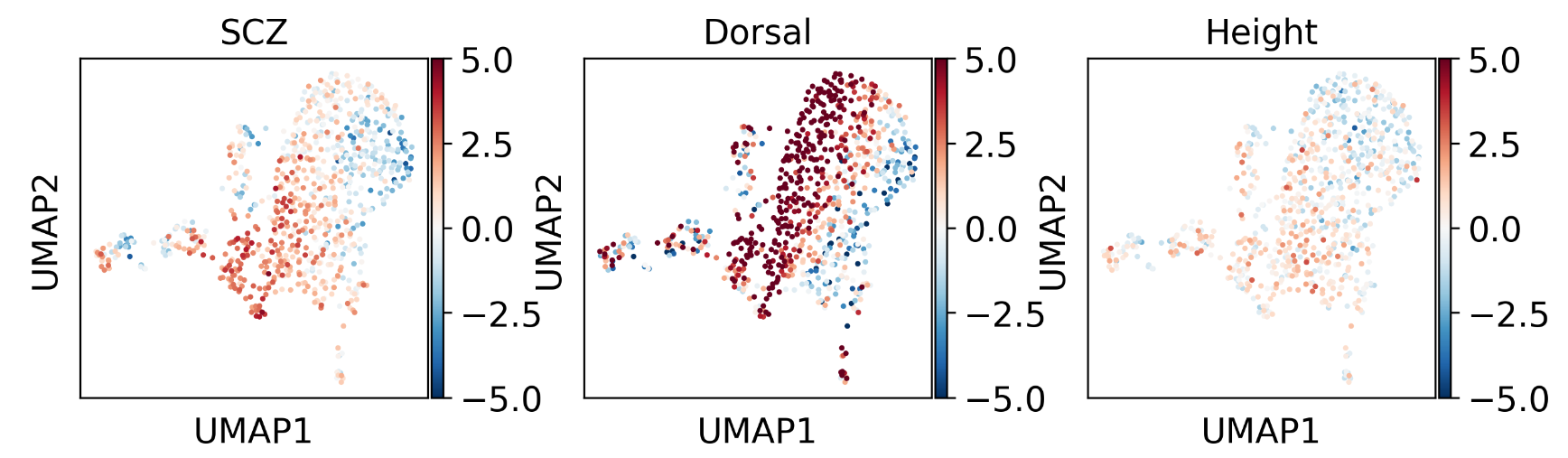
3.12 检测疾病与某类细胞的相关性水平
spatial_col = "Dorsal"
for trait in ["SCZ", "Height"]:
df_score = dict_score[trait].reindex(adata_ca1.obs.index)
ctrl_cols = [col for col in df_score.columns if col.startswith("ctrl_norm_score")]
n_ctrl = len(ctrl_cols)
# Pearson's r between trait score and spatial score
data_r = stats.pearsonr(df_score["norm_score"], adata_ca1.obs[spatial_col])[0]
# Regression: control score ~ spatial score
ctrl_r = np.zeros(len(ctrl_cols))
for ctrl_i, ctrl_col in enumerate(ctrl_cols):
ctrl_r[ctrl_i] = stats.pearsonr(df_score[ctrl_col], adata_ca1.obs[spatial_col])[
0
]
pval = (np.sum(data_r <= ctrl_r) + 1) / (n_ctrl + 1)
print(f"{trait} v.s. {spatial_col}, Pearson's r={data_r:.2g} (p={pval:.2g})")
结果如下所示:
SCZ v.s. Dorsal, Pearson's r=0.43 (p=0.001)
Height v.s. Dorsal, Pearson's r=0.04 (p=0.37)
致谢橙子牛奶糖(陈文燕),请用参考模版:We thank the blogger (orange_milk_sugar, Wenyan Chen) for XXX
感谢小可爱们多年来的陪伴, 我与你们一起成长~

本文来自博客园,作者:橙子牛奶糖(陈文燕),转载请注明原文链接:https://www.cnblogs.com/chenwenyan/p/16986027.html

