
shell实现碱基/氨基酸序列提取(sed,grep,awk三大利器走向天下)
假定存在file.fa文件,其序列如下所示:
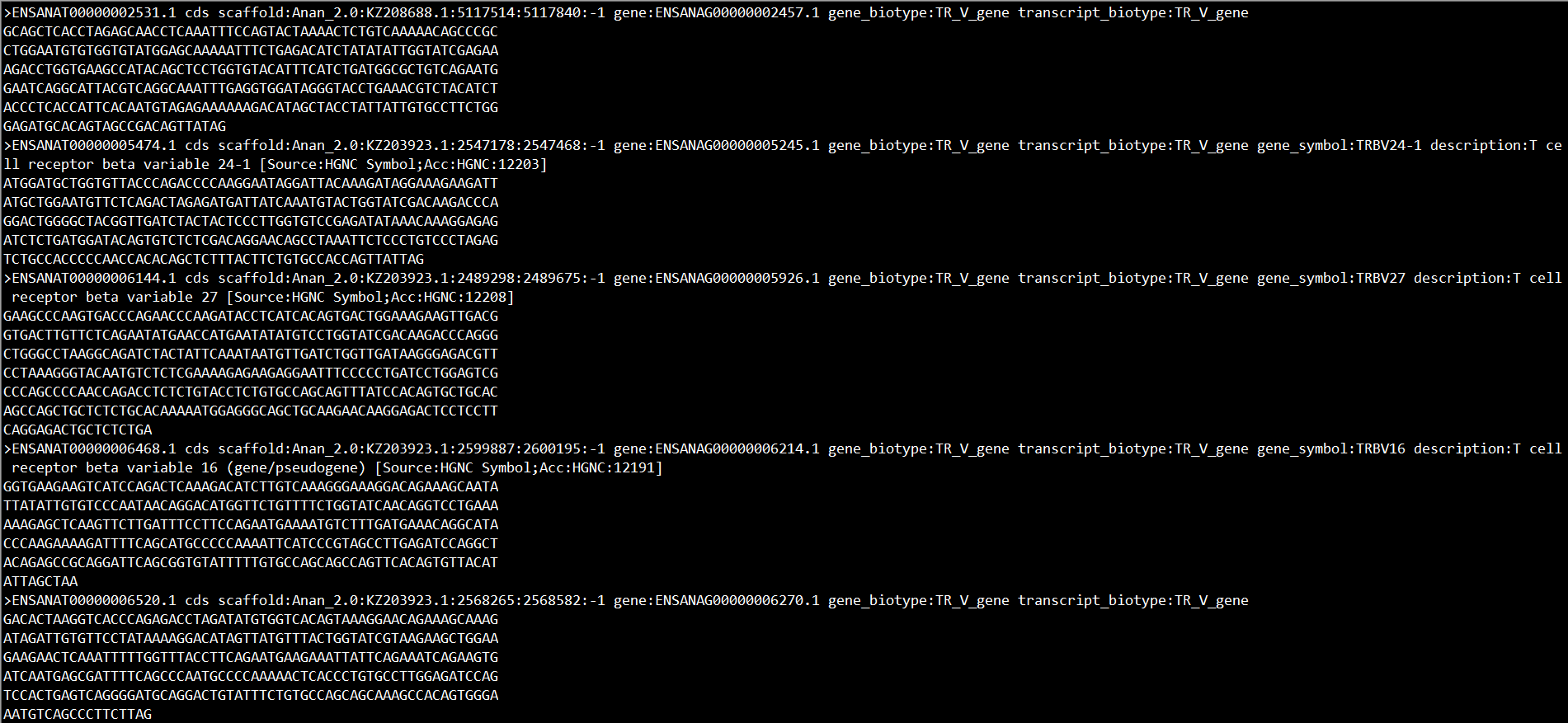
现在有四个任务,分别如下所示:
任务一:提取包含“RBFOX1”字符的序列
第一步:sed -i '/>/i\@' file.fa
file.fa指的是包含所有序列的文件;这一步的意思是在以">"开头的字符串前一行加上"@",方便后续序列的识别和匹配;
修改后的文件效果如下所示:

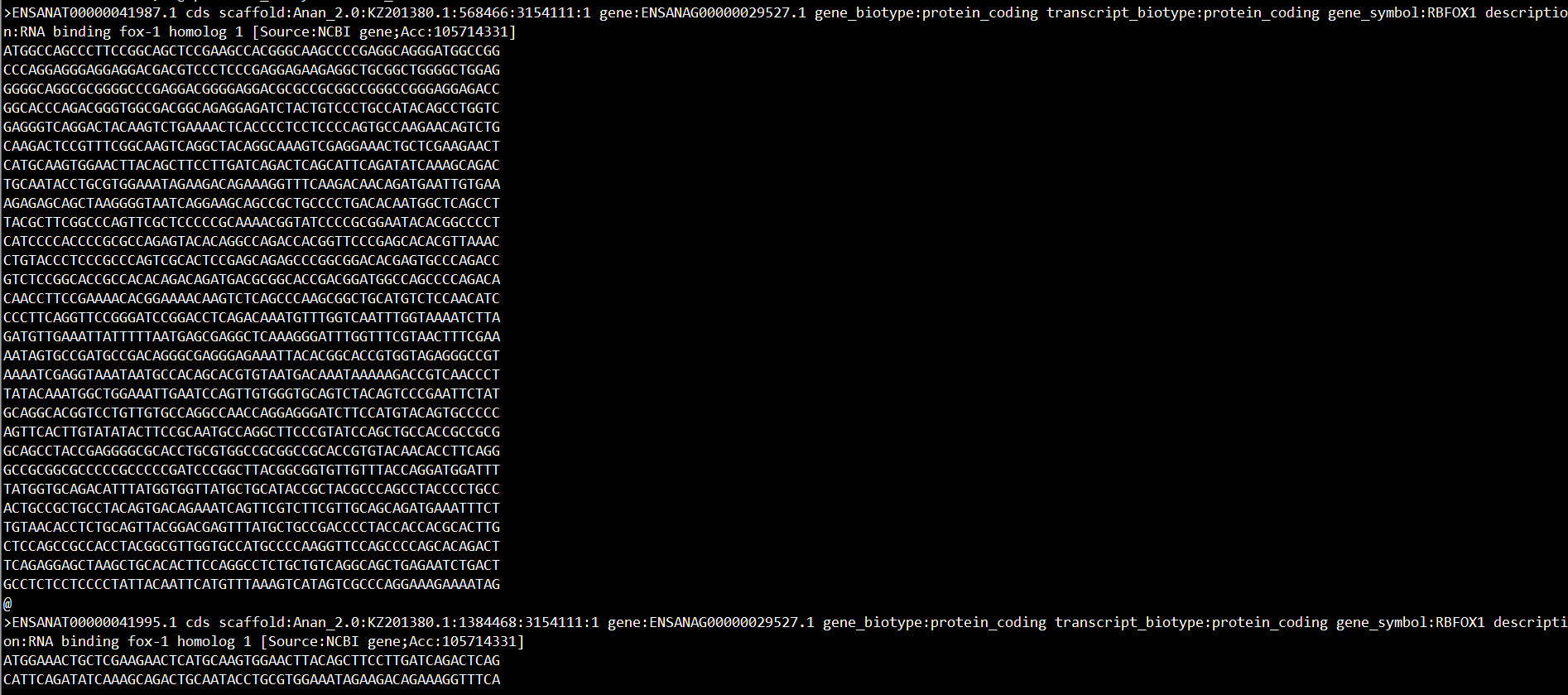
第二步:sed -n '/RBFOX1 /,/^@/p' file.fa > RBFOX1.fa
匹配“RBFOX1”和"@"字符的行,并打印包含“RBFOX1”和"@"字符之间的行
效果如下:
第三步:sed -i '/^@/d' RBFOX1.fa
删掉包含"@"所在的行
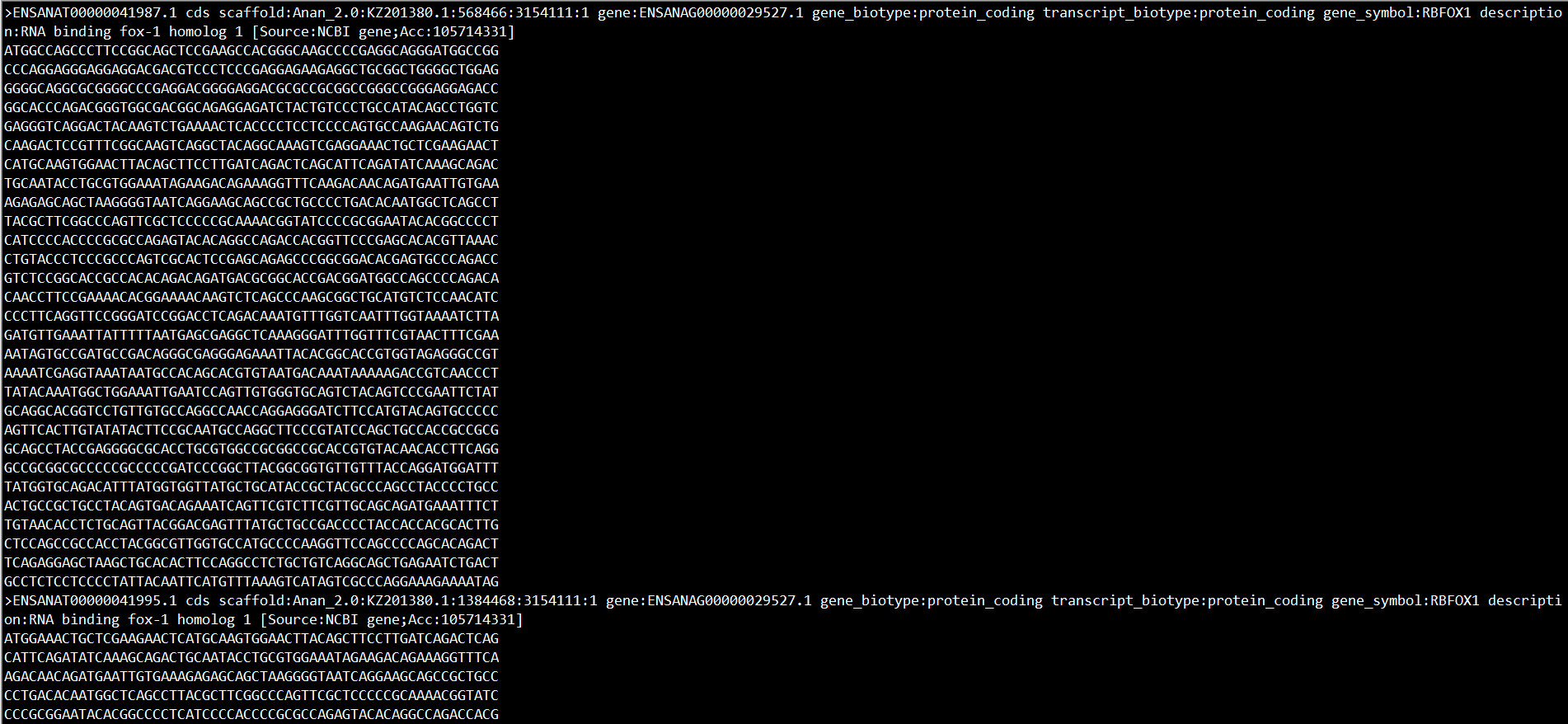
以上是提取RBFOX1序列的工作。
从上面可以看出,提取出来的RBFOX1序列不止一条,但很多时候我们只需要其中最长的一条,由此,引申了任务二。
任务二:从多条“RBFOX1”序列中提取最长的一段序列
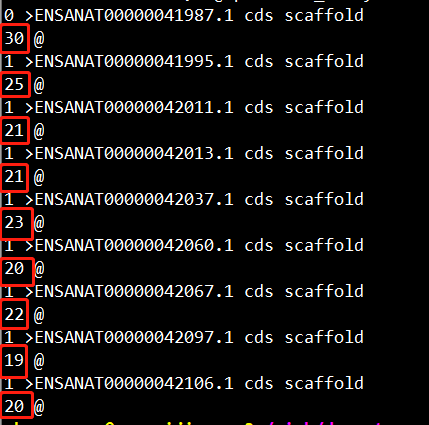
第一步:need1=`sed -n "/RBFOX1 /,/^@/p" file.fa | grep -En "@|>" | awk -F ":" 'NR==1{tmp=$1}NR>=0{print $1-tmp,$2;tmp=$1}'`
计算不同序列的长度,这里统计序列的行数(下图红色方框),行数越多,表明序列越长;
效果如下:
第二步:need2=`echo "$need1" | awk 'BEGIN {max = 0} {if ($1+0 > max+0) max=$1} END {print max}'`
统计最长的序列;
结果如下:

第三步:need3=`echo "$need1" | grep -B 1 "^$need2" | head -1 | awk '{print $2}'`
输出最长序列的ID;
结果如下:

第四步:提取最长序列
sed -n "/^$need3/,/^@/p" file.fa > RBFOX1.fa
sed -i '/^@/d' RBFOX1.fa
结果如下:

以上四步,即可完成从多条“RBFOX1”序列中提取最长序列的工作。
但在实际工作中,我们需要提取多个基因的序列,而非单个基因(比如只提取RBFOX1基因)的序列,由此引出任务三。
任务三:提取多个基因,且每条转录本的序列最长
首先,准备包含多个基因的文件genename,一个基因一行,其内容如下所示:
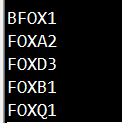
随后,输入如下命令:
while read line;do
need1=`sed -n "/${line} /,/^@/p" file.fa | grep -En "@|>" | awk -F ":" 'NR==1{tmp=$1}NR>=0{print $1-tmp,$2;tmp=$1}'`
need2=`echo "$need1" | awk 'BEGIN {max = 0} {if ($1+0 > max+0) max=$1} END {print max}'`
need3=`echo "$need1" | grep -B 1 "^$need2" | head -1 | awk '{print $2}'`
sed -n "/^$need3/,/^@/p" file.fa > ${line}.fa
sed -i '/^@/d' ${line}.fa
done < genename
以上命令即可一次性提取多个基因。
除了一次性提取多个基因外,我们还想一次性提取多个物种的多个基因,这个时候依旧可以使用循环命令完成工作。
任务四:提取多个物种、多个基因,且每条转录本的序列最长
假定有5个物种的序列存储在file.fa,file1.fa,file2.fa,file3.fa,file4.fa中,每一个fa文件代表一个物种。
此外,通过命令cat *.fa >> all.fa 生成 all.fa 文件,该文件包含5个物种的所有序列。
按照惯例,先在每个fa文件中加入"@",方便后续序列的识别和匹配,命令如下:
for j in *.fa; do
echo $j
sed -i '/>/i\@' $j
done
准备好以上文件后,输入如下命令即可完成多物种多基因,且每条转录本的序列最长的提取工作:
while read line;do
for i in file*.fa;do
need1=`sed -n "/${line} /,/^@/p" "$i" | grep -En "@|>" | awk -F ":" 'NR==1{tmp=$1}NR>=0{print $1-tmp,$2;tmp=$1} '`
need2=`echo "$need1" | awk 'BEGIN {max = 0} {if ($1+0 > max+0) max=$1} END {print max}'`
echo "$need1" | grep -B 1 "^$need2" | head -1 | awk '{print $2}' >> ${line}name
done
sed -i '/^\s*$/d' ${line}name
while read line1;do
echo ${line1}
sed -n -e "/^${line1}/,/^@/p" all.fa >> ${line}.fa
done < ${line}name
sed -i '/^@/d' ${line}.fa
done < genename
此推文感谢PCC同学的建议~

本文来自博客园,作者:橙子牛奶糖(陈文燕),转载请注明原文链接:https://www.cnblogs.com/chenwenyan/p/14341737.html

【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】凌霞软件回馈社区,博客园 & 1Panel & Halo 联合会员上线
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步