甲基化样本和CpG位点QC的总流程(450k和850k)
这篇应该是甲基化QC的最后一篇啦。
感谢健明带入门。
我前面已经写完两篇:
QC2:甲基化数据QC: 使用甲基化数据推测SNP基因型(ewastools工具)
下面补充一下对甲基化样本和CpG位点QC的总流程:
1、导入、加载安装包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GenomeInfoDbData")
BiocManager::install("IlluminaHumanMethylation450kmanifest")
BiocManager::install("IlluminaHumanMethylation450kanno.ilmn12.hg19")
BiocManager::install("IlluminaHumanMethylationEPICanno.ilm10b4.hg19")
BiocManager::install("IlluminaHumanMethylationEPICmanifest")
BiocManager::install("methylationArrayAnalysis")
BiocManager::install("limma")
BiocManager::install("minfi")
BiocManager::install("missMethyl")
BiocManager::install("minfiData")
BiocManager::install("Gviz")
BiocManager::install("DMRcate")
install.packages("knitr")
install.packages("RColorBrewer")
library(knitr)
library(limma)
library(minfi)
library(IlluminaHumanMethylationEPICanno.ilm10b4.hg19)
library(IlluminaHumanMethylation450kanno.ilmn12.hg19)
library(IlluminaHumanMethylation450kmanifest)
library(RColorBrewer)
library(missMethyl)
library(minfiData)
library(Gviz)
library(DMRcate)
library(stringr)
library("methylationArrayAnalysis")
2、加载数据
dataDirectory <- system.file("extdata", package = "methylationArrayAnalysis")
list.files(dataDirectory, recursive = TRUE)
3、加载甲基化注释包
ann450k <- getAnnotation(IlluminaHumanMethylation450kanno.ilmn12.hg19)
head(ann450k)
ann850k <- getAnnotation(IlluminaHumanMethylationEPICanno.ilm10b4.hg19)
head(ann850k)
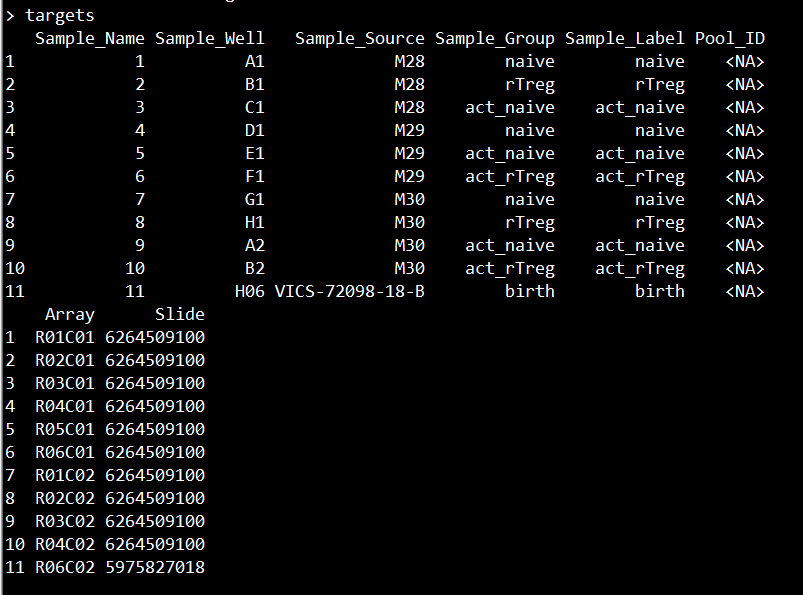
4、加载样本信息数据
targets <- read.metharray.sheet(dataDirectory, pattern="SampleSheet.csv")
targets
5、读取甲基化的原始数据idat
rgSet <- read.metharray.exp(targets=targets)
6、将样本名添加到甲基化数据中
targets$ID <- paste(targets$Sample_Group,targets$Sample_Name,sep=".")
sampleNames(rgSet) <- targets$ID
rgSet
开始QC~
7、甲基化cgp位点P值过滤
原理:对每一个样本的每一个Cpg位点的总信号(M+U)和背景信号进行比较,可以得到P值。一般认为,越低的P值表示该位点越可靠,P值大于0.01的cpg位点,是质量比较差的位点;
检测P值:
detP <- detectionP(rgSet)
head(detP)
结果如下图所示:
红色框框为样本名,蓝色框框为为一个cpg位点的P值。

画每个样本cpg位点的平均P值
pal <- brewer.pal(8,"Dark2")
par(mfrow=c(1,2))
barplot(colMeans(detP), col=pal[factor(targets$Sample_Group)], las=2,
cex.names=0.8, ylab="Mean detection p-values")
abline(h=0.05,col="red")
legend("topleft", legend=levels(factor(targets$Sample_Group)), fill=pal,
bg="white")
如下图所示:
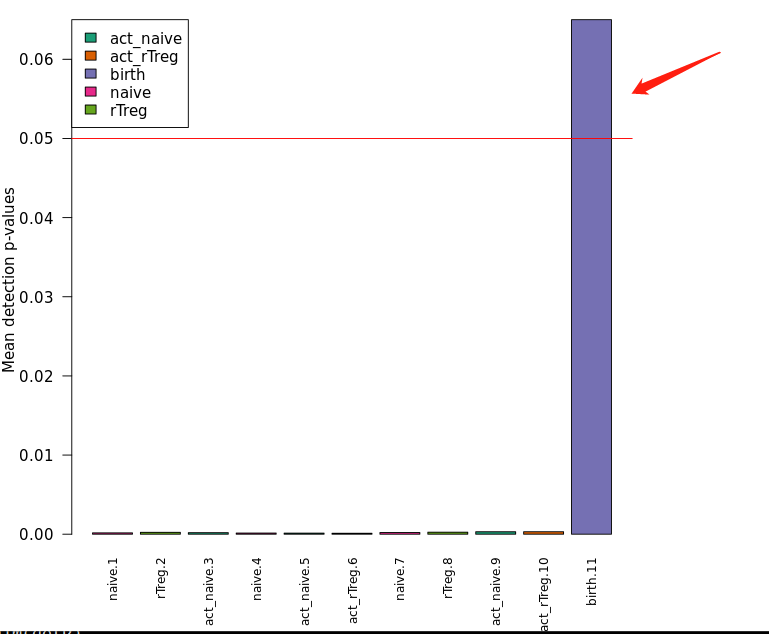
可以看到,只有最后一个样本的cpg平均P值是超过0.05,也就是说,这个样本的质量是比较差的,后续应该被剔除掉。
导出质量报道:
qcReport(rgSet, sampNames=targets\(ID, sampGroups=targets\)Sample_Group,
pdf="qcReport.pdf")
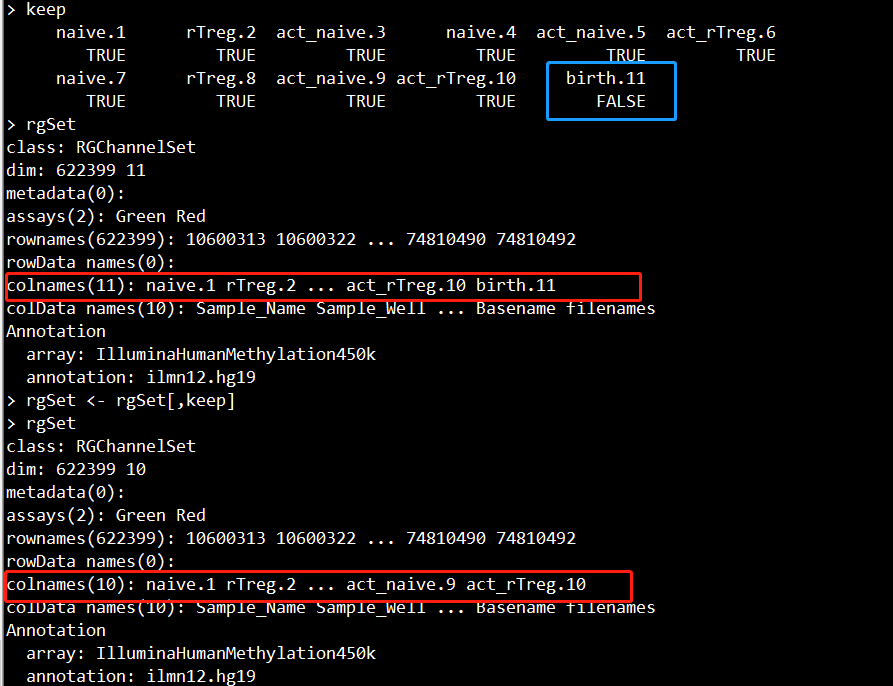
7.1、剔除甲基化中高P值样本
keep <- colMeans(detP) < 0.05
rgSet <- rgSet[,keep]
rgSet
这里对P值设定的阈值是大于0.05。
我们只保留cpg平均P值小于0.05的样本,对于P值大于0.05的样本(比如本例的birth.11)应被剔除。
剔除以后,11个样本就只剩下10个样本:
7.2、剔除样本信息中高P值的样本
targets <- targets[keep,]
targets[,1:5]
7.3、剔除P值中高P值的样本
detP <- detP[,keep]
dim(detP)
8、甲基化标准化
甲基化标准化是为了较少样本间的差异。
有两种包可以进行甲基化的标准化工作,分别为preprocessFunnorm和preprocessQuantile。
但这两个包的用途是不一样的。
preprocessFunnorm包是针对甲基化数据来源于有明显分层的样本,比如癌症样本和正常样本,皮肤组织样本和大脑组织的样本。像这种明显有不同来源的样本建议用preprocessFunnorm包进行标准化。
preprocessQuantile包则针对没有明显分层的样本,比如都是健康人群,都是来自血液样本这种情况。像这种单一来源的样本建议用preprocessQuantile包进行标准化。
使用preprocessQuantile包进行标准化:
mSetSq <- preprocessQuantile(rgSet)
比较标准化前后的样本的beta值分布
par(mfrow=c(1,2))
densityPlot(rgSet, sampGroups=targets$Sample_Group,main="Raw", legend=FALSE)
legend("top", legend = levels(factor(targets$Sample_Group)),
text.col=brewer.pal(8,"Dark2"))
densityPlot(getBeta(mSetSq), sampGroups=targets$Sample_Group,
main="Normalized", legend=FALSE)
legend("top", legend = levels(factor(targets$Sample_Group)),
text.col=brewer.pal(8,"Dark2"))
画出来的图如下所示,左边是未进行标准化的,右边是标准化以后的。
可见,进行标准化后,样本间的差异会缩小。

9、查找标准化后数据可能存在的差异来源
这一步是为EWAS做准备的,我们前面进行了标准化,但标准化的数据不代表就可以完全去除样本批次效应、细胞类型等差异。
如果样本间存在批次效应等可能的混淆因素,在后续进行EWAS分析时极大可能会产生假阳性。
因此我们需要通过主成分分析对标准化的数据进行可视化。确定可能的混淆因素,并在EWAS分析时进行校正。
10、通过主成分1、2确认样本间的差异来源
par(mfrow=c(1,2))
plotMDS(getM(mSetSq), top=1000, gene.selection="common",
col=pal[factor(targets$Sample_Group)])
legend("top", legend=levels(factor(targets$Sample_Group)), text.col=pal,
bg="white", cex=0.7)
plotMDS(getM(mSetSq), top=1000, gene.selection="common",
col=pal[factor(targets$Sample_Source)])
legend("top", legend=levels(factor(targets$Sample_Source)), text.col=pal,
bg="white", cex=0.7)
如下图所示,可以很明显的看到这10个样本被分为三个聚类。
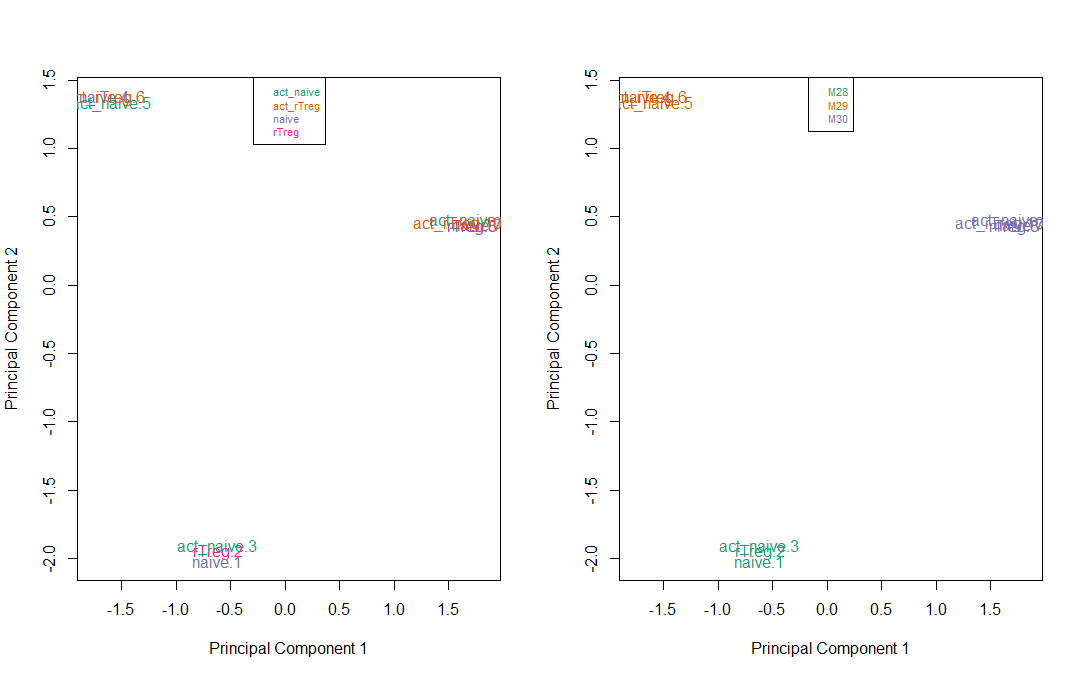
说明即便是前期进行了标准化处理后,样本间还是存在差异,比如样本act_naive.5和naive.1就很明显的在不同的聚类中。
这种差异在进行EWAS分析时是我们不愿意看到的,因此后期进行EWAS分析时,应考虑将他们纳入协变量中。
11、通过主成分1、2、3、4确认样本间的其他差异来源
par(mfrow=c(1,3))
plotMDS(getM(mSetSq), top=1000, gene.selection="common",
col=pal[factor(targets$Sample_Group)], dim=c(1,3))
legend("top", legend=levels(factor(targets$Sample_Group)), text.col=pal,
cex=0.7, bg="white")
plotMDS(getM(mSetSq), top=1000, gene.selection="common",
col=pal[factor(targets$Sample_Group)], dim=c(2,3))
legend("topleft", legend=levels(factor(targets$Sample_Group)), text.col=pal,
cex=0.7, bg="white")
plotMDS(getM(mSetSq), top=1000, gene.selection="common",
col=pal[factor(targets$Sample_Group)], dim=c(3,4))
legend("topright", legend=levels(factor(targets$Sample_Group)), text.col=pal,
cex=0.7, bg="white")
对主成分1,2,3,4画图后如下所示:
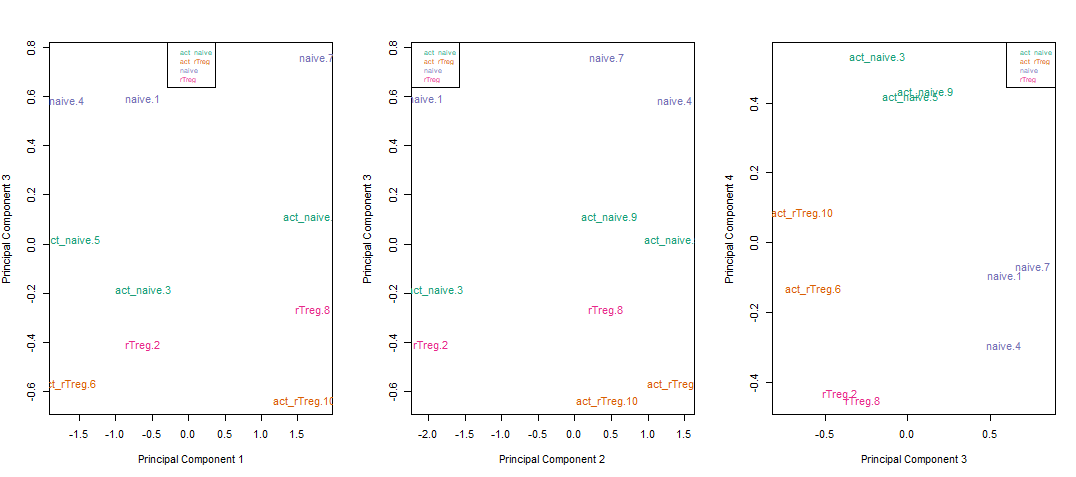
可以看到,不同样本按照不同颜色很好地被分层了,说明主成分3,4反应的是细胞类型的差异。
同样的,细胞类型差异在进行EWAS分析时也是我们并不愿意看到的,因此他们在进行EWAS分析时应被一起纳入协变量中校正掉。
12、探针过滤
前面我们根据cpg的P值结果对样本进行了过滤。
现在我们需要对探针进行过滤。
detP <- detP[match(featureNames(mSetSq),rownames(detP)),] #匹配ID
keep <- rowSums(detP < 0.01) == ncol(mSetSq) #对P值小于0.01的探针进行计数
table(keep) #统计有多少个探针P值小于0.01
mSetSqFlt <- mSetSq[keep,] #保留P值在所有样本中均小于0.01的探针。
mSetSqFlt
13、移除包含性染色体的探针
keep <- !(featureNames(mSetSqFlt) %in% ann450k$Name[ann450k$chr %in% c("chrX","chrY")])
table(keep)
mSetSqFlt <- mSetSqFlt[keep,]
14、移除SNP探针
mSetSqFlt <- dropLociWithSnps(mSetSqFlt)
mSetSqFlt
15.1、移除匹配在多个基因组上的探针(如果是450k)
这一步是针对450k的数据,如果你的数据是850k,略过这一步,请看下面850k的工作。
xReactiveProbes <- read.csv(file=paste(dataDirectory,
"48639-non-specific-probes-Illumina450k.csv",
sep="/"), stringsAsFactors=FALSE)
keep <- !(featureNames(mSetSqFlt) %in% xReactiveProbes$TargetID)
table(keep)
mSetSqFlt <- mSetSqFlt[keep,]
mSetSqFlt
15.2移除匹配在多个基因组上的探针(如果是850k)
这一步是针对850k的数据,如果你的数据是450k,略过这一步,请看上面450k的工作。
if (! ("devtools" %in% installed.packages()) install.packages("devtools")
devtools::install_github("markgene/maxprobes")
library(maxprobes)
xloci <- maxprobes::xreactive_probes(array_type = "EPIC")
length(xloci)
mSetSqFlt <- maxprobes::dropXreactiveLoci(mSetSqFlt)
16.1、重新评估是否已经消除样本间的差异(方法一:minfi包)
par(mfrow=c(1,2))
plotMDS(getM(mSetSqFlt), top=1000, gene.selection="common",
col=pal[factor(targets$Sample_Group)], cex=0.8)
legend("right", legend=levels(factor(targets$Sample_Group)), text.col=pal,
cex=0.65, bg="white")
plotMDS(getM(mSetSqFlt), top=1000, gene.selection="common",
col=pal[factor(targets$Sample_Source)])
legend("right", legend=levels(factor(targets$Sample_Source)), text.col=pal,
cex=0.7, bg="white")

par(mfrow=c(1,3))
plotMDS(getM(mSetSqFlt), top=1000, gene.selection="common",
col=pal[factor(targets$Sample_Source)], dim=c(1,3))
legend("right", legend=levels(factor(targets$Sample_Source)), text.col=pal,
cex=0.7, bg="white")
plotMDS(getM(mSetSqFlt), top=1000, gene.selection="common",
col=pal[factor(targets$Sample_Source)], dim=c(2,3))
legend("topright", legend=levels(factor(targets$Sample_Source)), text.col=pal,
cex=0.7, bg="white")
plotMDS(getM(mSetSqFlt), top=1000, gene.selection="common",
col=pal[factor(targets$Sample_Source)], dim=c(3,4))
legend("right", legend=levels(factor(targets$Sample_Source)), text.col=pal,
cex=0.7, bg="white")

在这里,我们可以看到样本的组别、来源差异相比未标准化和未过滤前,已经减少了很多。
16.2、重新评估是否已经消除样本间的差异(方法二:ChAMP包)
bVals <- getBeta(mSetSqFlt)
champ.SVD(beta = bVals ,
rgSet=NULL,
pd=targets,
RGEffect=FALSE,
PDFplot=TRUE,
Rplot=TRUE,
resultsDir="./CHAMP_SVDimages/")

计算原理是:先对甲基化beta值做主成分分析,对每个主成分和变量(比如本例中的sample_label,sample_group,sample_source,ID,Array等)进行kruskal.test检验,确定两组或多组的中位数是否存在差异。
如果存在差异,说明变量和甲基化beta值存在相关性,也就是说,变量不能被很好的校正掉,那么,将这些没有被很好校正掉的甲基化数值进行后续分析的话,就很容易产生假阳性。
从上面截图可以看到,sample_label,sample_group,sample_source这几个变量与甲基化主成分显著相关,后续做EWAS分析时应将他们作为协变量纳入分析中,或者用ChAMP包的champ.runCombat函数或者minfi包的sva函数将这些变量进行校正。
17、提取M值和beta值
提取M值(mVals)和beta值(bVals):
mVals <- getM(mSetSqFlt)
head(mVals[,1:5])
bVals <- getBeta(mSetSqFlt)
head(bVals[,1:5])
对M值(mVals)和beta值(bVals)进行画图:
par(mfrow=c(1,2))
densityPlot(bVals, sampGroups=targets$Sample_Group, main="Beta values",
legend=FALSE, xlab="Beta values")
legend("top", legend = levels(factor(targets$Sample_Group)),
text.col=brewer.pal(8,"Dark2"))
densityPlot(mVals, sampGroups=targets$Sample_Group, main="M-values",
legend=FALSE, xlab="M values")
legend("topleft", legend = levels(factor(targets$Sample_Group)),
text.col=brewer.pal(8,"Dark2"))
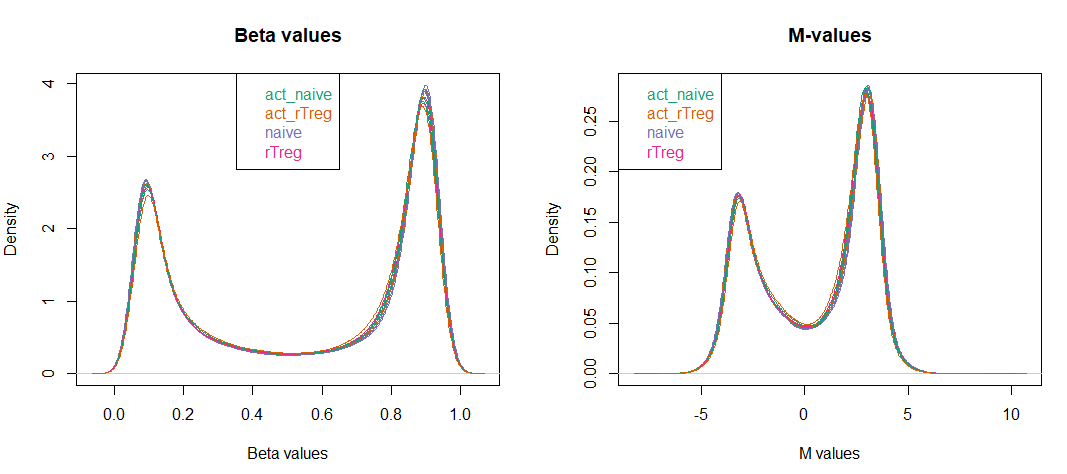
收获美美的双峰!
上面的教程大部分是基于minfi包展开的。
实际上,除了minfi包,ChAMP包也可以完成这个工作,ChAMP包更简单。直接四个函数搞定。
如下截图所示。
这里我就不展开讲了,原理跟minfi包一样的,只不过ChAMP包把它封装好了。
champ.filter(beta=myImport$beta,
M=NULL,
pd=myImport$pd,
intensity=NULL,
Meth=NULL,
UnMeth=NULL,
detP=NULL,
beadcount=NULL,
autoimpute=TRUE,
filterDetP=TRUE,
ProbeCutoff=0,
SampleCutoff=0.1,
detPcut=0.01,
filterBeads=TRUE,
beadCutoff=0.05,
filterNoCG = TRUE,
filterSNPs = TRUE,
population = NULL,
filterMultiHit = TRUE,
filterXY = TRUE,
fixOutlier = TRUE,
arraytype = "EPIC")
champ.QC(beta = myLoad$beta,
pheno=myLoad$pd$Sample_Group,
mdsPlot=TRUE,
densityPlot=TRUE,
dendrogram=TRUE,
PDFplot=TRUE,
Rplot=TRUE,
Feature.sel="None",
resultsDir="./CHAMP_QCimages/")
champ.norm(beta=myLoad$beta,
rgSet=myLoad$rgSet,
mset=myLoad$mset,
resultsDir="./CHAMP_Normalization/",
method="BMIQ",
plotBMIQ=FALSE,
arraytype="EPIC",
cores=3)
champ.SVD(beta = myNorm,
rgSet=NULL,
pd=myLoad$pd,
RGEffect=FALSE,
PDFplot=TRUE,
Rplot=TRUE,
resultsDir="./CHAMP_SVDimages/")
甲基化QC工作到此结束啦~
18、总结
minfi流程多、繁琐,胜在轻巧,按着流程走,一般不会出现什么报错。
ChAMP包方便,但如果数据多的话,对电脑的配置要求也很高,我跑3000个样本时,256G,32cpu核是带不动的,经常跑着跑着就被kill了。用几百个样本跑时,就很顺利。
19、致谢
感谢健明分享的甲基化分析入门练习:甲基化芯片的一般分析流程
建议各位刚入门甲基化的同学们可以看看健明在B站的视频,讲的很详细。

本文来自博客园,作者:橙子牛奶糖(陈文燕),转载请注明原文链接:https://www.cnblogs.com/chenwenyan/p/13184835.html

 浙公网安备 33010602011771号
浙公网安备 33010602011771号