甲基化数据QC: 使用甲基化数据推测SNP基因型(ewastools工具)
介绍一下如何使用ewastools推测甲基化数据的SNP基因型
下载、安装ewastools
以下例子是ewastools的安装,其他的包比如magrittr、svd等, 电脑没装的话,也需要一一装上去。
如果install.packages()装不了,就用BiocManager::install()安装。
生物信息的R包,用这两个跑不了。
install.packages("devtools")
library("devtools")
devtools::install_github("hhhh5/ewastools@master")
library(ewastools)
library(stringi)
library(data.table)
library(magrittr)
library(purrr)
library(svd)
library("dplyr")
导入数据
这里依旧用的是minfi的示例文件。
先下载、安装minfi包:
BiocManager::install(c("minfi","minfiData","sva"))
library(minfi)
library(minfiData)
library(sva)
导入甲基化数据:
baseDir <- system.file("extdata", package="minfiData")
targets <- read.metharray.sheet(baseDir)
RGSet <- read.metharray.exp(targets = targets)
manifest <- getManifest(RGSet)
manifest
甲基化的数据如下,我们可以看到,这里是有65个SNP探针位点的。
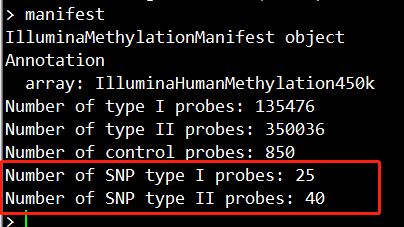
提取甲基化数据中的SNP
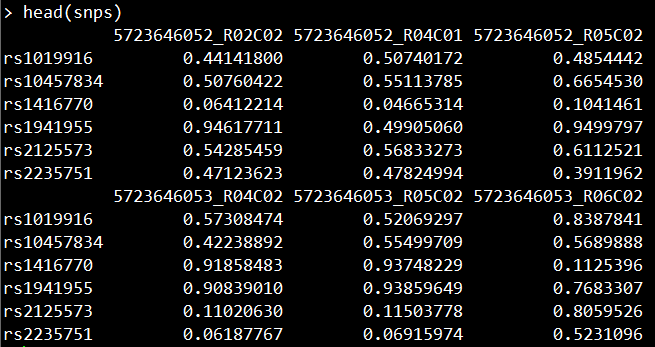
使用minfi包的getSnpBeta函数提取甲基化数据中的SNP位点以及对应的数值。
snps <- getSnpBeta(RGSet)
head(snps)
对甲基化数据call SNP的基因型
将上面一步提取出来的甲基化SNP位点进行基因型的calling。
使用ewastools包的call_genotypes函数:
genotypes = call_genotypes(snps,learn=FALSE)
提取推测的基因型:
ge3=data.frame(genotypes[3])
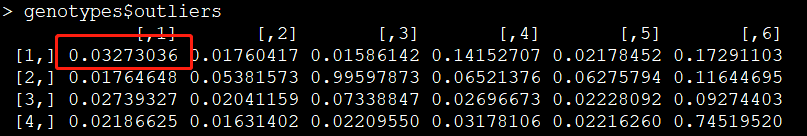
提取异常值:
outliners=genotypes$outliers
结果解读
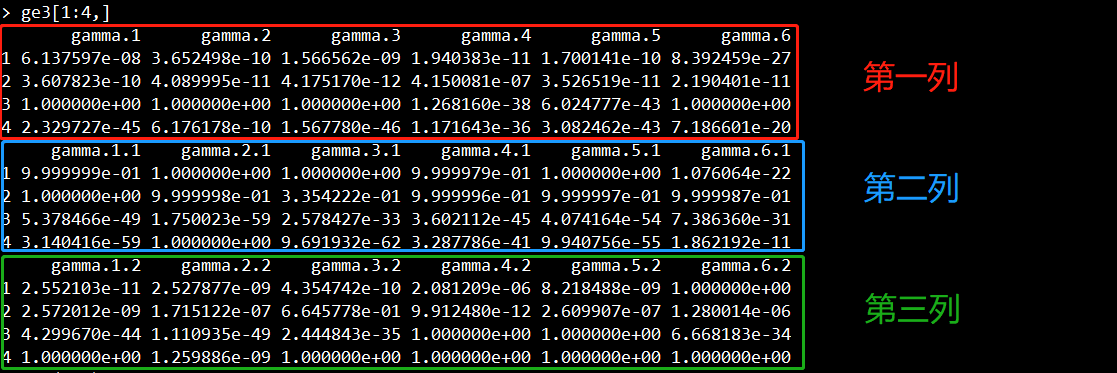
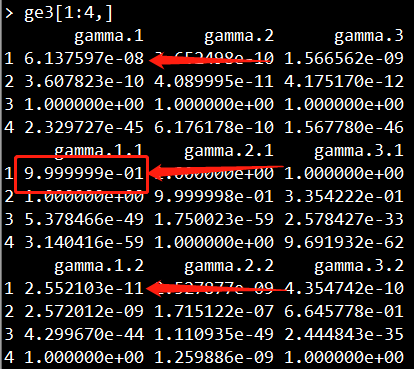
结果如下所示,每一行表示一个SNP,每一列表示一个样本。所以这里显示有6个样本,4个SNP。
每个样本的每个SNP会call出三种不同的分布(分别对应截图显示的第一列、第二列、第三列)。
三种分布对应着三种基因类型:minor纯合子、杂合型、major纯合子。
如果第一列的值大于第二列、第三列、异常值,那么该位点为纯合子;
如果第二列大于第一列、第三列、异常值,那么该位点为杂合子;
如果第三列的值大于第一列、第二列、异常值,那么该位点为纯合子;
如果该位点的异常值大于第一列、第二列、第三列,那么该位点的基因型无法推测;
第一列和第三列哪个对应的是minor纯合子和major纯合子,则需要进行统计计算。
第一列、第二列、第三列示意图:
异常值示意图:
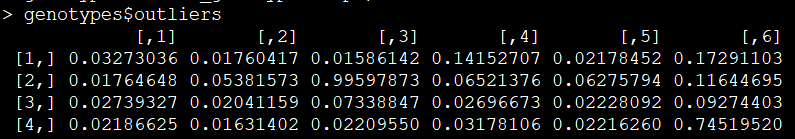
举个例子,如下两张截图所示,对于第一个样本的第一个位点,其三个分布的值为6.137597e-08、9.999999e-01、2.552103e-11,异常值为0.03273036。显然第二列的值最大,因此该样本在该位点的基因型为杂合子。

本文来自博客园,作者:橙子牛奶糖(陈文燕),转载请注明原文链接:https://www.cnblogs.com/chenwenyan/p/13031425.html

 浙公网安备 33010602011771号
浙公网安备 33010602011771号