作业要求:
在UCSC下载hg19参考基因组,我博客有详细说明,从gencode数据库下载基因注释文件,并且用IGV去查看你感兴趣的基因的结构,比如TP53,KRAS,EGFR等等。
作业,截图几个基因的IGV可视化结构!还可以下载ENSEMBL,NCBI的gtf,也导入IGV看看,截图基因结构。了解IGV常识。
参考基因组--下载
地址:UCSC https://genome.ucsc.edu/

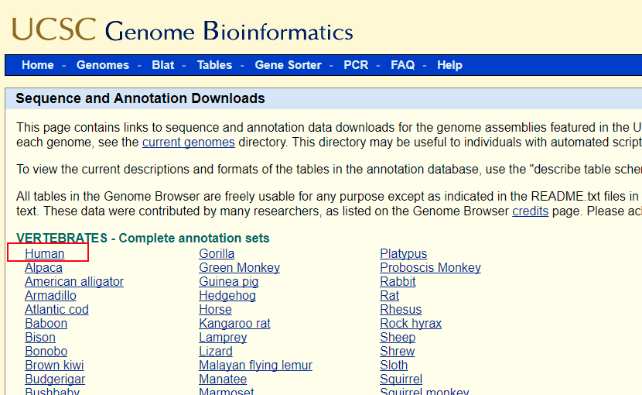
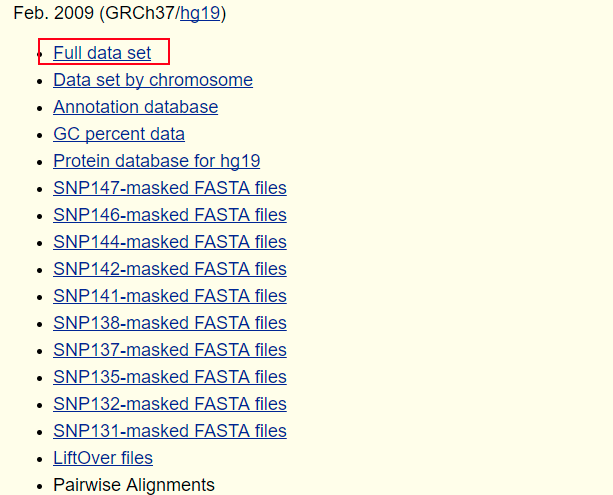
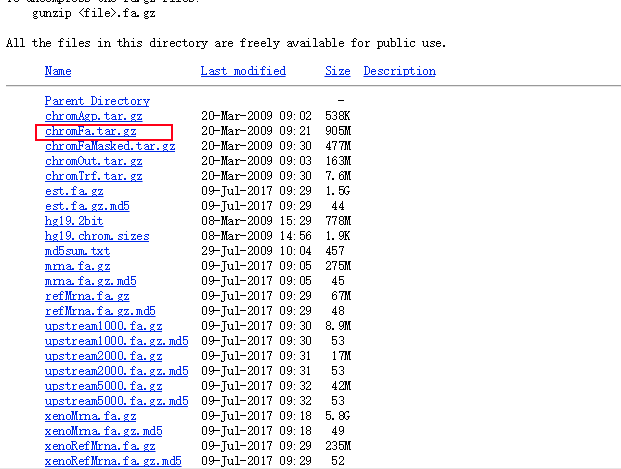
(1)、进入UCSC---选择Downloads---genomic data---human---GRCh37/hg19---Full data set,下拉,找到chromFa.tar.gz,右击chromFa.tar.gz,选择“复制链接地址”
# 点击 Full data set后,有各类文件的说明文档
(2)、终端命令行操作
1 # 切换到要存放参考基因组的目录 2 $ cd data/GSE81916/reference/genome/hg19 3 4 # 用axel或wget下载参考基因组 5 $ nohup wget http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz # wget后面跟的是参考基因组的下载地址 6 7 # 解压下载后的文件 8 $ tar -zxvf chromFa.tar.gz 9 10 # 解压后可以发现,参考序列是按照染色体号分开列出的,我们还需要把所有的序列写入到一个文件中。 11 $ cat *.fa > hg19.fa 12 13 #最后删除其他无用的文件 14 $ rm chr*.fa
注释文件--下载
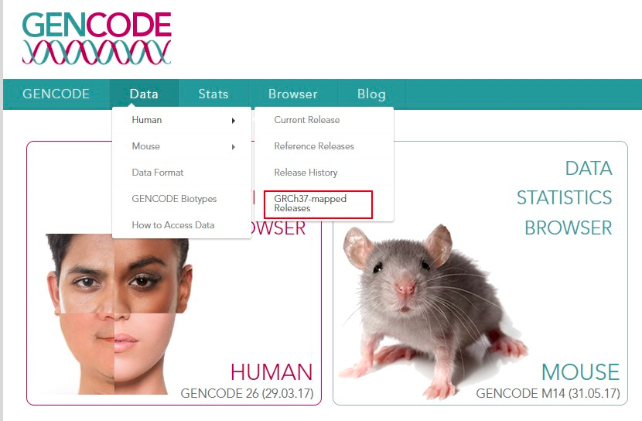
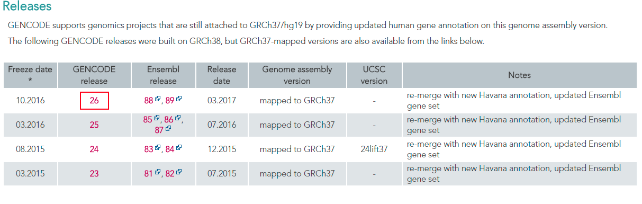

(1)、进入Gencode数据库---Data---Human---GRCh37-mapped Release---选择2016年10月份发布的最新注释版本“gencode.v26lift37.annotation.gtf.gz”
鼠标右击,“复制链接地址”
(2)、命令行批量下载
1 # 用axel批量下载 2 $ axel ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v26lift37.annotation.gtf.gz 3 # 下载后解压 4 $ gzip -d gencode.v26lift37.annotation.gtf.gz 5 # 与下载的hg19参考基因组放在一起 6 $ mv genconde.v26lift37.annotation.gtf ./Reference/Human/hg19
补充:GTF和GFF之间的区别
数据结构:都是由9列构成,分别是reference sequence name; annotation source; feature type; start coordinate; end coordinate; score; strand; frame; attributes.前8列都是相同的,第9列不同。
GFF第9列:都是以键值对的形式,键值之间用“=”连接,不同属性之间用“;”分隔,都是以ID这个属性开始。下图中有两个ID,说明是不同的序列。

GTF第9列:同样以键值对的形式,键值之间是以空格区分,值用双引号括起来;不同属性之间用“;”分隔;开头必须是geneid, transciptid两个属性。

基因组浏览器:IGV
Integrative Genomics Viewer(IGV)是一种探索大型综合基因组数据的高性能交互式可视化工具。它支持各种各样的数据类型,包括基于芯片测序、二代测序数据和基因组注释数据等。
IGV下载
1 # 进入IGV官网,并下载相应的软件包,有Windows,Mac,和LINUX,这里我下载Linux二进制包 2 $ cd ~/src 3 $ wget http://data.broadinstitute.org/igv/projects/downloads/IGV_2.3.97.zip 4 $ unzip IGV_2.3.97.zip && mv IGV_2.3.97 ~/biosoft 5 6 # 添加环境变量 7 $ vim ~/.bashrc 8 PATH=$PATH:~/biosoft/IGV_2.3.97 9 $ source ~/.bashrc 10 11 # 运行IGV,Linux直接运行igv.sh可以开启IGV窗口,但是会比较慢,要耐心等待。 12 $ igv.sh
IGV使用
0、初始化窗口
1、载入基因组,选择Genome标签,load我们之前已经下载好的hg19.fa基因组。
2、载入基因组注释,但是在载入之前需要将gff3进行排序,选择Tools-Run igvtools,进入以下igvtools窗口:
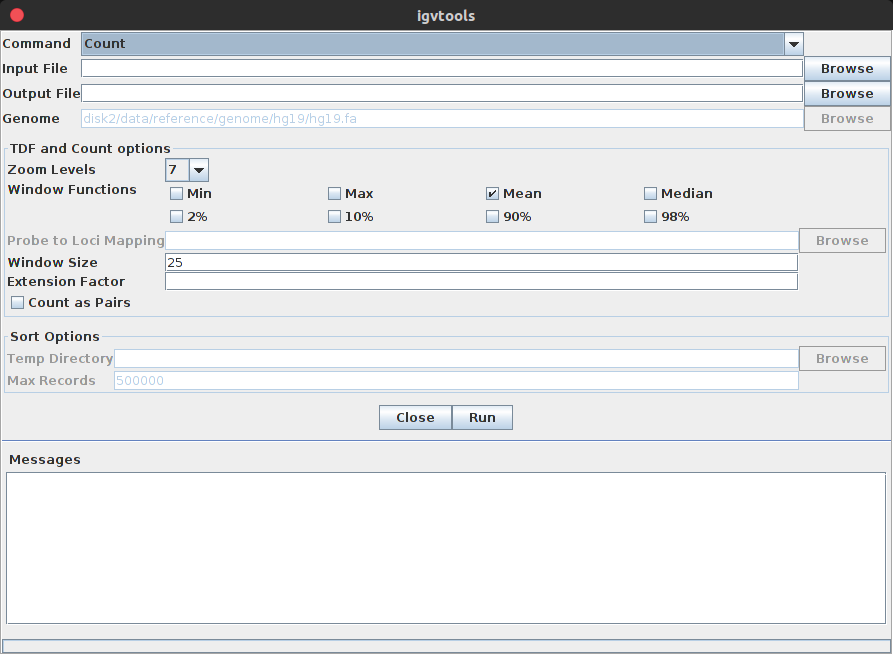
3、获得sorted文件:command选择sort,再选择输入的注释文件,点击Run,就可以生成sorted.gff3文件。
4、通过file->load from file...选择sorted文件,打开。选择区域的大小,来看某些基因的信息,蓝色的粗线条就是代表基因。说到底,IGV就是一个将基因组及其注释信息可视化的工具。 (下图是载入基因组和注释信息后的窗口)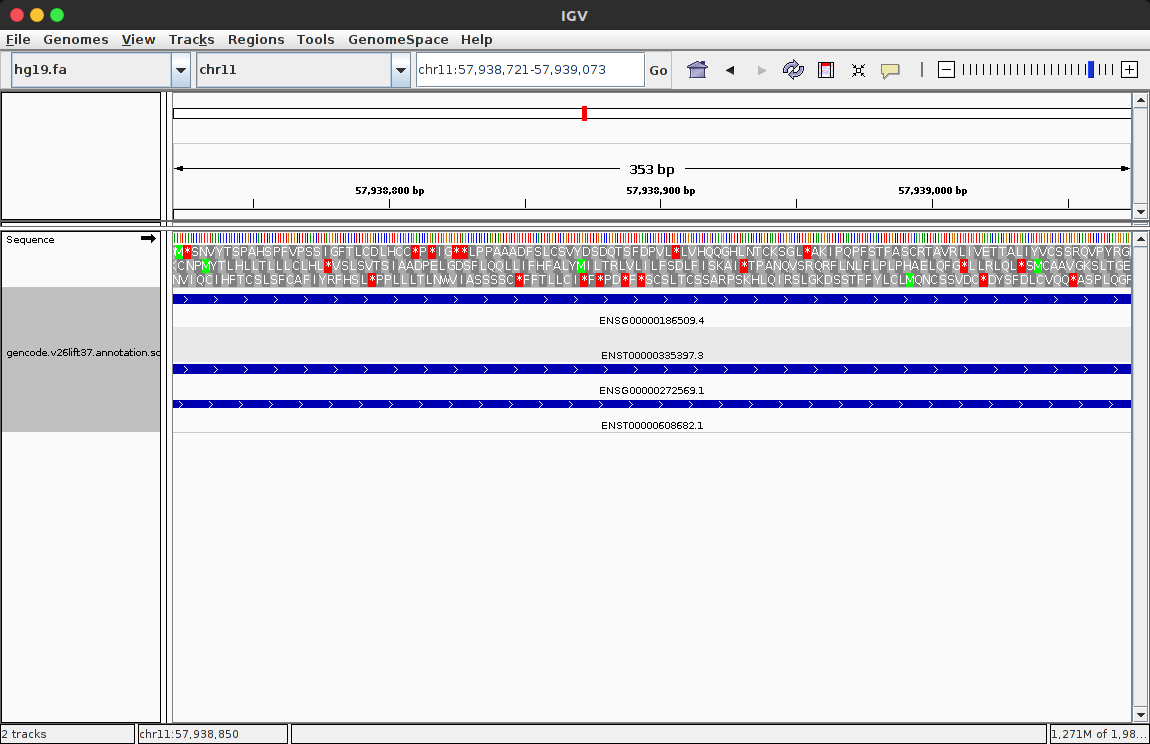
理论知识
RNA-seq数据分析的通用套路是:
1、检测测序数据的质量,如果需要,对数据进行预处理,去掉接头,去掉质量差的数据等等
2、将所有数据回帖到genome,根据结果,进行新基因或转录本的鉴定,然后对转录数据进行定量,并进行差异表达分析。也可跳过对新基因和新转录本的分析,只对已知的基因和转录本进行定量。
3、如果没有参考genome数据,可以供transcritome数据代替。
4、如果参考转录组数据也没有,可以直接对RNA-seq数据进行从头组装,注释,作为参考转录组。
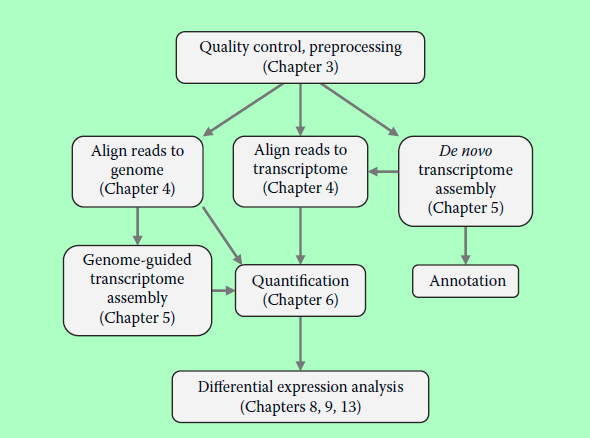
图片源于《RNA-seq Data Analysis》
把高通量测序得到的reads回帖到参考基因组上,既是进行后续基因表达定量和差异表达分析的基础,同时也是可变剪接分析、SNP、InDel分析以及测试数据质量控制的一部分
回帖常用软件:HISAT2, STAR
参考基因组
参考基因组的实质:就是某一物种的基因组序列,因此是fasta格式。
三大网站:
1.NCBI (https://www.ncbi.nlm.nih.gov/grc)
2.UCSC (http://hgdownload.soe.ucsc.edu/downloads.html)
3.Ensemble (http://asia.ensembl.org/index.html?redirect=no)
三大网站的ftp地址:
ensembl : ftp://ftp.ensembl.org/pub
NCBI : ftp://ftp.ncbi.nih.gov/genomes/
UCSC:ftp://hgdownload.soe.ucsc.edu/goldenPath
推荐:去Ensemble下载参考序列,(UCSC很久没更新)
目前最常用的人的参考基因组版本如下(Jimmy总结):
|
NCBI |
UCSC |
Ensemble |
|
GRCh36 |
hg18 |
ENSEMBL release_52 |
|
GRCh37 |
hg19 |
ENSEMBL release_59/61/64/68/69/75 |
|
GRCh38 |
hg38 |
ENSEMBL release_76/77/78/80/81/8 |
注释文件
就是基因组的说明书。告诉我们哪些序列是编码蛋白的基因,哪些是非编码基因,外显子、内含子、UTR等的位置等等。注释文件在以上三个提供参考基因组的网站中都有提供,比如Ensemble。但是现在最权威的人类和小鼠基因组的注释还属Gencode数据库。
IGV软件界面简介
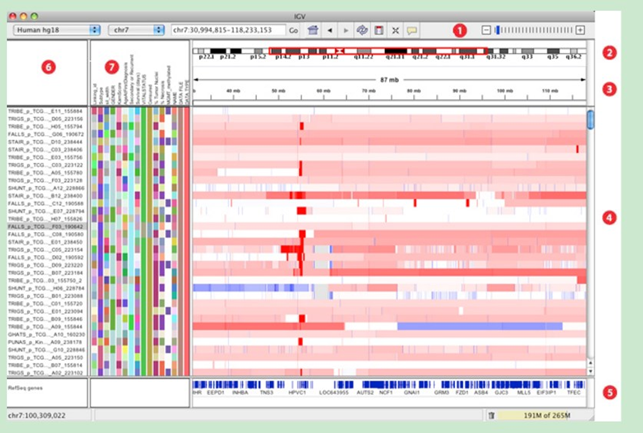
主窗口布局:
1.工具栏tool bar
2.红色框显示当前显示的染色体的位置,当缩小显示范围到整个染色体范围时,红色框消失。
3.显示当前查看的染色体序列的长度
4.该窗口显示测序样品的测序情况。每一条track代表一个样品或者一次实验,显示的情况包括甲基化、表达水平、拷贝数,碱基突变等信息。
5.参考基因组信息
6.track名(即样品或者实验名)
7.Attribute names属性名,即序列信息,如indel、甲基化等。
更多的使用方法可查看IGV User Guide
参考资料
转录组入门(1)-作业-转录组-生信技能树 http://www.biotrainee.com/thread-1796-1-1.html
HOPTOP转录组入门(一)布置运行环境-转录组-生信技能树 http://www.biotrainee.com/thread-1800-1-1.html
RNA-seq基础入门传送门-转录组-生信技能树 http://www.biotrainee.com/thread-1750-1-1.html
浙大植物学小白的转录组笔记 http://www.360doc.com/content/17/0911/22/46164085_686360709.shtml

 浙公网安备 33010602011771号
浙公网安备 33010602011771号