计算机资源准备
操作系统:mac、Linux(双系统或虚拟机)
配置:8G+内存,500G存储
配置举例:
| 内存 | 8G,DRR3-1600-16芯的4G内存条 2根 |
| SSD | 120G,特科芯 |
| HDD | 1T,希捷5200转 |
| CPU | P9600 |
需要安装的软件:sratoolkit, fastqc, hisats, samtools, htseq-count, R, Rstudio
ubuntu操作系统准备
1 # 备份源文件,然后将默认镜像源改成清华源 2 $ 3 # 更新升级索引 4 $ sudo apt-get update 5 $ sudo apt-get upgrade
# 创建软件下载目录src和软件目录biosoft(用于编译安装)
$ cd && mkdir src && mkdir biosoft
anaconda
简介:软件管家,快速便捷的安装软件,推荐
参考:
https://zhuanlan.zhihu.com/p/25085567
http://python.jobbole.com/86236/
sratoolkit
作用:数据格式转换
介绍: sratoolkit的主要用途还是把NCBI SRA(Sequence Read Archive)数据库中的NGS序列数据从 sra 格式转换到 fastq 格式,以便于后续的数据分析。
功能:下载,操作,验证NCBI SRA中二代测序数据
提高:1、如何开启ascp加速下载;2、vdb-config更改基本设置
官网主页:https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software
中文相关介绍一篇:http://www.cnblogs.com/OA-maque/p/4799074.html
安装与测试:
#安装 #用anaconda安装 conda install -c jfear sratoolkit
#编译安装:
# 切换到软件下载目录 $ cd && cd src
# 选择适合自己系统的版本,Ubuntu
$ wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.8.2-1/sratoolkit.2.8.2-1-ubuntu64.tar.gz
# 解压并将解压后的文件剪切到biosoft目录下 $ tar -zxvf sratoolkit.2.8.2-1-ubuntu64.tar.gz
$ mv sratoolkit.2.8.2-1-ubuntu64 ~/biosoft # vim编辑器直接编辑~/.bashrc文件,将该软件加入环境变量中,可以全局运行,不用在运行的时候切换到当前目录
$ vim ~/.bashrc
# 在文件最后增加内容: PATH=$PATH:~/biosoft/sratoolkit.2.8.2-1-ubuntu64/bin
# 更新
$ source ~/.bashrc
# 测试 prefetch -v # 下载测试文件SRR390728,默认存放在家目录下的ncbi文件夹中 prefetch -c SRR390728
fastqc
作用:分析测序数据质量
介绍:二代测序数据质量分析软件
功能:可视化展示二代测序数据质量
提高:1、理解每个图的含义;2、掌握如何从fastqc的结果图中获取信息;
3、掌握sed用法http://dongweiming.github.io/sed_and_awk/
官网主页:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
中文相关介绍一篇:https://zhuanlan.zhihu.com/p/20731723
安装与测试:
1 # anaconda安装 2 $ conda install fastqc 3 4 # 编译安装 5 # 看是否安装了Java(fastqc运行需要Java环境) 6 $ java -version 7 # 若不存在,则进行安装,但是Java的版本要适合。我在装了Java9之后,fastqc没法正常运行,之后降到8版本之后,就能正常运行。 8 $ sudo apt-get openjdk-8-jdk 9 $ cd ~/src 10 # 下载二进制包,对自己Linux有信心的同志,可以下载源码包,自己编译 11 $ wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.5.zip 12 $ unzip fastqc_v0.11.5.zip && mv FastQC ~/biosoft 13 $ vim ~/.bashrc 14 # 加入环境变量 15 PATH=$PATH:~/biosoft/FastQC 16 $ source ~/.bashrc 17 18 19 # 测试 20 $ fastqc -h # 测试,出现帮助信息 21 $ fastqc #测试是否能正常打开该软件
hisat2
简介:将测序结果比对(回帖)到人类参考基因组上。
背景:HISAT2是TopHat2/Bowti2的继任者,使用改进的BWT算法,实现了更快的速度和更少的资源占用。
官网主页:http://ccb.jhu.edu/software/hisat2/index.shtml
中文相关介绍:https://www.plob.org/article/10380.html
安装与测试:
1 # anaconda安装 2 $ conda install hisat2 3 4 # 编译安装 5 # 切换到下载目录 6 $ cd ~/src 7 # 直接下载二进制包,免去自己进行编译安装 8 $ wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads/hisat2-2.1.0-Linux_x86_64.zip 9 $ unzip hisat2-2.1.0-Linux_x86_64.zip && mv hisat2-2.1.0 ~/biosoft 10 # 添加环境变量 11 $ vim ~/.bashrc 12 # 在文件最后加入:PATH=$PATH:~/biosoft/hisat2-2.1.0 13 # 更新 14 $ source ~/.bashrc 15 16 17 # 测试 18 $ hisat2 -h # 出现帮助信息,说明安装成功
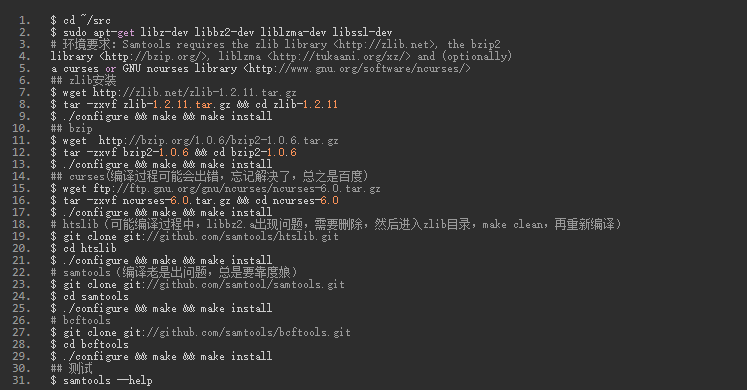
samtools
作用:文件操作
介绍:一种处理SAM、BAM文件的工具软件。BAM格式文件是存放高通量测序中比对结果的标准格式文件。
功能:Reading/writing/editing/indexing/viewing SAM/BAM/CRAM format
官网主页:旧主页http://samtools.sourceforge.net/ 新主页http://www.htslib.org
中文相关介绍:http://www.cnblogs.com/OA-maque/p/4827146.html
安装与测试:
# 安装 $ conda install samtools # 安装2(如果上面的方法不行)
$ ocnda install -c bioconda samtools=1.8
# 测试 $ samtools –help
htseq-count
简介:计数
功能:根据比对结果统计基因count
介绍:htseq-count 是一款用于reads计数的软件,他能对位于基因组上的一些单位的reads数进行统计,这里所说的单位主要是指染色体上的一组位置区间(我们常见的就是gene exon)
官网主页:http://www-huber.embl.de/users/anders/HTSeq/doc/install.html#installation-on-linux
中文相关介绍:http://yangl.net/2016/09/21/htseq-count_manual/
http://www.cnblogs.com/OA-maque/p/4835033.html
安装与测试:
# 安装 #anaconda 只能安装0.7.2版没有最新版 $ conda install -c bioconda htseq # 编译安装 ubuntu $ sudo apt-get install build-essential python2.7-dev python-numpy python-matplotlib # 编译安装 Redhat系列 包括CentOs $ sudo yum install python-devel numpy python-matplotlib #下载HTSeq $ wget [url=https://pypi.python.org/packages]https://pypi.python.org/packages[/url] ... /HTSeq-0.8.0.tar.gz # 解压 $ tar -zxvf HTSeq-0.8.0.tar.gz # 安装 $ mv HTSeq-0.8.0 Biosoft #移动到Biosoft文件夹中 $ cd HTSeq-0.8.0 #进入该文件夹 $ python setup.py install –user #安装 # 测试 在非HTSeq-0.8.0文件夹下进行 $ python >>>import HTSeq >>> #能够在python中导入HTSeq这个包,说明安装成功。
R
作用:统计,画图,后续分析
简介:一种常用语统计分析的编程语言,在生物信息分析中用于数据分析和绘图
官网主页:https://www.r-project.org
中文相关介绍:http://www.oschina.net/p/r-language
安装与测试:
# 安装(方法1) # 1、添加镜像源到/etc/apt/sources.list(注意Ubuntu版本) $ sudo vim /etc/apt/sources.list # 把 deb http://cran.rstudio.com/bin/linux/ubuntu xenial/ 粘贴到文件末尾 # 2、下载公钥 $ gpg --keyserver keyserver.ubuntu.com --recv-key 51716619E084DAB9 $ gpg -a --export 51716619E084DAB9 | sudo apt-key add -12 # 3、安装 r-base $ sudo apt-get update $ sudo apt-get install r-base r-base-dev
# 安装(方法2)(推荐试试!)
# 打开source.list文件,添加中国科学技术大学的镜像源
$ vim /etc/apt/source.list
# 添加如下内容
deb https://mirrors.ustc.edu.cn/CRAN/bin/linux/ubuntu xenial/
deb https://mirrors.ustc.edu.cn/ubuntu/ xenial-backports main restricted universe
# 添加密钥到电脑,secoure APT
$ sudo apt-key adv --keyserver keyserver.ubuntu.com --recv-keys E084DAB9
$ sudo apt-get update
$ sudo apt-get install r-base
$ sudo apt-get install r-base-dev
# 安装(方法3)
# 添加Secure APTs
sudo apt-key adv --keyserver keyserver.ubuntu.com --recv-keys E084DAB9 # 添加deb到source.list vi source.list deb [url=https://mirrors.ustc.edu.cn/CRAN/bin/linux/ubuntu]https://mirrors.ustc.edu.cn/CRAN/bin/linux/ubuntu[/url] xenial/ deb [url=https://mirrors.ustc.edu.cn/ubuntu/]https://mirrors.ustc.edu.cn/ubuntu/[/url] xenial-backports main restricted universe # 更新并安装 sudo apt-get updatesudo apt-get install r-base # (optional)如果要自己编译R sudo apt-get install r-base-dev
# 测试
$ R
# (可选)提高R包安装速度,可以再家目录下新建.Rprofile,添加如下内容
$ vim /.Rprofile
options("repos" = c(CRAN = "https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
options(BioC_mirror = "https://mirrors.tuna.tsinghua.edu.cn/bioconductor")
Rstudio
简介:Rstudio是R的集成开发环境,很好用的IDE
官网主页:https://www.rstudio.com
中文相关介绍:http://jingyan.baidu.com/article/a17d52853343828098c8f21c.html
安装与测试:
# conda安装 $ conda install rstudio
# 编译安装
# 切换到下载目录
$ cd ~/src
$ wget https://download.rstudio.org/rstudio-1.0.143-amd64.deb
# 安装,可以直接点击deb包, # 测试 $ rstudio
参考资料
转录组入门(1)-作业-转录组-生信技能树 http://www.biotrainee.com/thread-1796-1-1.html
HOPTOP转录组入门(一)布置运行环境-转录组-生信技能树 http://www.biotrainee.com/thread-1800-1-1.html
RNA-seq基础入门传送门-转录组-生信技能树 http://www.biotrainee.com/thread-1750-1-1.html
浙大植物学小白的转录组笔记 http://www.360doc.com/content/17/0911/22/46164085_686360709.shtml

 浙公网安备 33010602011771号
浙公网安备 33010602011771号