生物医学顶刊论文(JBHI-2024):TransFOL:药物相互作用中复杂关系推理的逻辑查询模型
(2024.5.17)JBHI-TransFOL:药物相互作用中复杂关系推理的逻辑查询模型
论文题目:TransFOL: A Logical Query Model for Complex Relational Reasoning in Drug-Drug Interaction
论文期刊:Journal of Biomedical and Health Informatics (JBHI)
论文地址:https://ieeexplore.ieee.org/document/10530338/
摘要
预测药物-药物相互作用(DDI)在药物推荐和发现中扮演着至关重要的角色。然而,由于药物相互作用,实验室方法的成本昂贵且耗时。近年来,深度学习方法在药物推理中得到了广泛应用。尽管这些方法已经证明了其有效性,它们只能预测药物对之间的相互作用,并不包含其他任何信息。然而,DDI在很大程度上受到各种其他生物医学因素的影响(例如药物剂量)。因此,将它们应用于更复杂和有意义的推理任务是具有挑战性的。因此,本研究将DDI视为知识图谱上的链接预测问题,并提出了一个基于Cross-Transformer和图卷积网络(GCN)的一阶逻辑查询形式的DDI预测模型,TransFOL。在模型中,首先构建生物医学查询图以学习嵌入表示。随后,设计了一个增强模块来聚合实体和关系的含义。交叉变换器用于编码以获取节点之间的语义信息,图卷积网络用于进一步收集邻居信息并预测推理结果。为了评估TransFOL在常见DDI任务上的性能,我们在两个基准数据集上进行了实验。实验结果表明,我们的模型在传统DDI任务上的性能超过了最先进的方法。此外,我们在其他两个实验中引入了不同的生物医学信息,以使设置更加现实。实验结果验证了TransFOL在复杂设置中强大的药物推理能力和泛化能力。数据和代码可在 https://github.com/Cheng0829/TransFOL 上获取。
1.引言
药物相互作用(DDI)预测对于药物重新定位、药物推荐和药物发现至关重要,药物功能的未知变化可能导致不良药物反应。预测药物、疾病和蛋白质等因素之间的潜在相互作用对于避免严重的副作用至关重要。尽管临床试验可以发现不良反应,但某些严重的不良反应只有在患者接受治疗时才被发现。检测和预防不良反应需要大量的人力和财力资源,并且实现安全可靠的临床试验具有挑战性。
因此,提出了自动分析方法来解决这一问题。然而,传统的机器学习模型通常需要手动特征工程,这需要专业知识并且耗时费力。
最近的研究调查了贝叶斯方法和深度学习模型。例如,Takeda等人考虑了药物相互作用预测中的药代动力学(PK)或药效动力学(PD)副作用,并开发了一种结合分子相似性和药物副作用相似性的贝叶斯网络用于药物效果预测。CNN-DDI使用卷积神经网络来学习药物相互作用预测的特征表示。然而,这些方法仅将药物-药物、药物-靶标和药物-表型相互作用视为外部知识,可能对于复杂的生物医学任务不够全面。知识图谱(KG)和知识表示学习方法也促进了药物相互作用预测领域的应用。MCFF-MTDDI通过有效融合药物化学结构特征、药物对的附加标签特征和药物的KG特征,以及使用门控循环单元的多通道特征融合模块,解决了特征冗余和KG噪声问题。
随着生物医学知识图谱的出现,更多类型的实体和关系被应用于这一任务。TriVec模型是一种KG嵌入模型,通过神经网络将实体及其关系投射到稠密向量进行预测。STNN-DDI提出了一种子结构感知张量模型,将药物映射到子结构空间,能够以统一且可解释的方式执行多类型的药物相互作用预测。然而,大多数方法在推理过程中未能直接利用目标实体的邻域信息,使得模型无法充分利用实际的外部知识。更重要的是,它们只能预测最简单的药物对之间的相互作用,无法扩展到更复杂的场景,限制了它们在现实世界中的应用。
在这种背景下,本研究将药物相互作用问题框架化为知识图谱上的链路预测任务,并提出了一种名为TransFOL的药物相互作用预测模型,该模型基于GCN和Cross-Transformer,旨在应对一阶逻辑查询框架内独特的复杂关系推理需求。关系推理指的是通过分析实体之间的关联和属性信息推断潜在的相互作用或依赖性。在制药领域的复杂关系推理具体指需要考虑多个生物医学因素(如药物剂量、代谢途径和疾病背景)或能够处理多跳因果链、条件约束和非线性效应的推理任务。这种复杂性源于药物相互作用不仅受到药物直接配对的影响,还受到其他生物医学因素的综合调控。TransFOL旨在解决传统方法难以有效处理的复杂推理问题,从而提高药物相互作用预测的准确性和实用性。
在TransFOL的模型架构中,三重转换将知识图谱转化为没有边缘属性的有向查询图。我们进一步使用即插即用的双向聚合功能增强实体和关系的语义表示。然后,我们采用Cross-Transformer和GCN解码器来收集邻域信息并预测药物相互作用。
从具体任务角度来看,我们在四个任务中全面测试了TransFOL的推理能力。在多类别DDI预测任务(任务1)中,我们预测两种药物之间的唯一相互作用类型;在多标签DDI预测任务(任务2)中,我们同时预测多种相互作用类型。
除了上述常见的简单药物相互作用预测任务外,我们还研究了带有额外剂量属性信息的复杂预测。例如,在任务3中,我们将药物剂量信息引入药物相互作用预测。具有属性信息的预测更准确和现实,对现有预测方法来说具有挑战性。这表明TransFOL在复杂环境中的药物知识发现能力更强。
此外,我们设计了一个更复杂且有价值的任务(任务4),要求模型仅基于疾病信息和哺乳动物临床症状推断人类临床症状,这有助于解决当前药物开发过程中临床周期过长的劣势,展示了TransFOL的泛化能力。
总之,这项工作引入了TransFOL模型,这是一种通过逻辑查询推理进行药物知识发现的新方法。TransFOL在通过逻辑查询表达生物医学实体之间复杂相互作用方面表现突出。在多个基准数据集上的多个任务中表现优异,并优于多个强基线。
2.预备知识
设 \(\mathcal{G}=(\mathcal{E},\mathcal{R})\) 表示一个知识图谱(KG),其中 \(e \in \mathcal{E}\) 表示一个实体,\(r \in \mathcal{R}\) 是一个二元谓词(或关系)\(r: \mathcal{E} \times \mathcal{E} \rightarrow \{\mathrm{True, False}\}\),用于指示某对实体之间的关系是否成立。给定一阶逻辑中的存在量词(\(\exists\))和合取运算(\(\wedge\)),合取查询定义如下:
其中 \(A\) 指的是查询的(未知的)目标实体,\(E_1, \cdots, E_m\) 指的是存在量化的绑定变量(即未知的中间实体集合),而 \(c\) 指的是源实体。给定查询 \(\mathcal{Q}\),目标是找到满足 \(a \in \mathcal{A}\) 当且仅当 \(\mathcal{Q}[a]\) 为真时的目标实体集合 \(\mathcal{A} \subseteq \mathcal{E}\)。
在这里,我们用一个简单的例子来解释逻辑查询是如何工作的。要将问题 "阿片类镇痛药能治疗哪些疾病?" 转换成逻辑查询,我们需要在特定约束下进行2跳推理,即遵循一系列步骤(用普通语言)来获得查询答案。首先,我们搜索阿片类药物实体,即第一个约束。然后,我们寻找能够缓解疼痛的药物实体,即与前一个相同的第二个约束。最后,在第二跳推理中找到由所获得实体治疗的疾病。这个逻辑查询是一个常见的"\(2p\)"型查询,它使用2跳知识图谱查询。通过分析逻辑查询,我们可以看到查询的第二跳提供了我们所需的答案,即所获得实体治疗的疾病。附录I提供了知识图谱分析中常用的逻辑查询类型的更多示例。
除了合取查询(\(\wedge\)),一阶逻辑(FOL)查询还包括析取查询(\(\vee\))。基于离散数学的知识,我们知道一个析取标准形式可以分解成几个合取典型形式(如附录II所示),从而可以应用规则来综合析取预测的合取结果。换句话说,所有正一阶逻辑查询都可以转换为典型的析取形式进行统一处理。
3.方法
本节介绍了TransFOL,这是一种基于Cross-Transformer和图卷积网络(GCN)的一阶逻辑查询模型,用于解决生物医学知识图谱上的DDI预测问题。为了使TransFOL能够有效地聚合复杂的语义和邻居信息,我们提出使用Cross-Transformer作为编码器,并在编码前后采用聚合函数增强模块和GCN解码器作为进一步增强。
在3.1节中,我们总结了TransFOL的整体结构。3.2节描述了三元组转换策略,它能够使异构图转换为无边属性的有向逻辑查询图。3.3节介绍了一个即插即用的表示增强模块。3.4节提出了一个融合模型,该模型使用Cross-Transformer和GCN作为骨干神经网络,并利用知识图谱的结构和与药物相关的知识信息来进一步学习药物的表示。
3.1 模型概括
TransFOL的框架如图1所示。我们的模型包括四个模块:三元组转换、表示增强模块、Cross-Transformer和GCN解码器。
首先,为了实现实体和关系的统一建模,我们采用三元组转换操作,将关系边转换为关系节点,将知识图谱(KG)转换为无边属性的有向图。我们在三元组转换模块中将知识图谱三元组转换为一种多实体元组。具体来说,我们将关系转换为关系节点,并将知识图谱转换为无边属性的有向图。我们采用这种方法将知识图中的实体关系转换为逻辑查询形式,并将边属性信息转换为节点表示,使得DDI预测任务可以在统一的节点嵌入空间中处理,同时利用生物医学实体关系的典型稀疏性质确保计算效率。
在三元组转换操作之后,我们将知识图谱转换为异构图查询。然后,我们使用表示增强模块来聚合语义信息。我们采用即插即用的双向注意力机制来整合实体、关系和类型的表示,使中间变量节点能够感知锚点节点或目标节点的信息以及推理链中的相关信息,避免冗余或不相关信息的干扰。这有助于防止由于长链推理而削弱锚点节点与目标实体之间的连接,使模型能够更好地理解和捕获实体之间的相互作用。
接下来,在交叉变换器模块中,我们设计了一个双层策略,包括药物特征分离和融合层。在药物特征分离层中,我们分别对实体和关系进行编码。在药物特征融合层中,我们融合特征,然后使用扁平化操作来学习全局特征。这使模型能够在通过特定的交叉注意力机制实现跨实体类型语义传播的同时,捕获更丰富的实体和关系特征。
最后,在GCN模块中,模型通过卷积操作从多跳邻居中提取有价值信息,从而聚合局部邻居信息。
模型采用Cross-Transformer和GCN的双层架构。这种设计有效地分离和整合不同特征,学习全局上下文,增强了模型推断复杂DDI的能力。
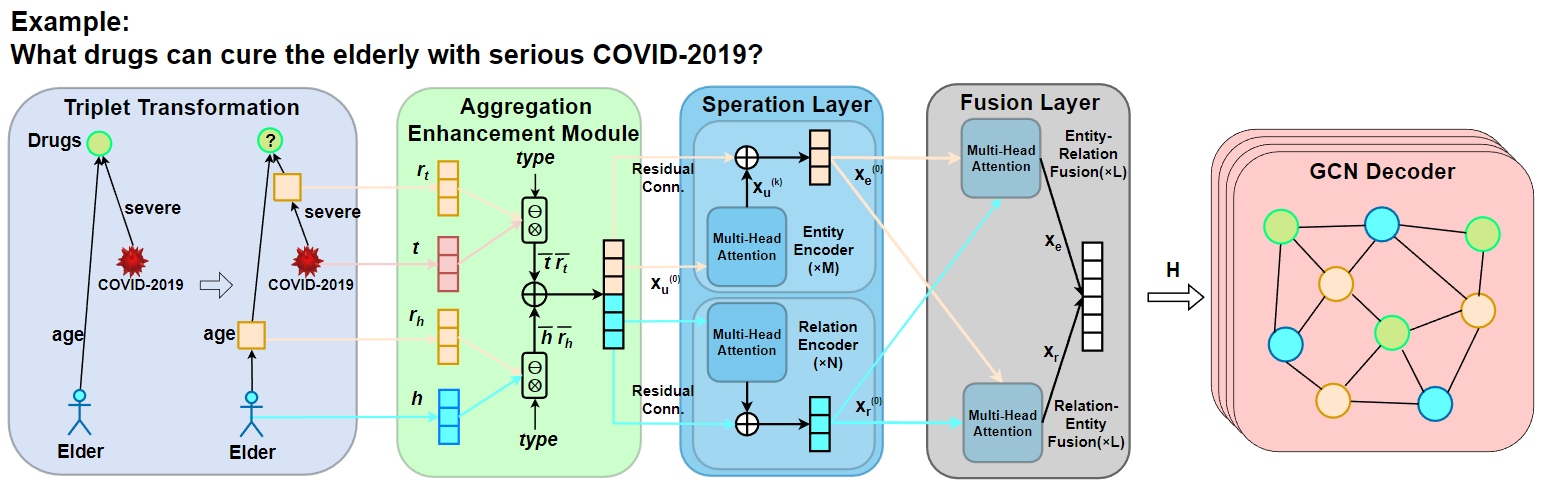
图1:TransFOL模型的总体结构。(a) 三元组转换模块将实体的属性信息作为逻辑查询图中的关系来处理。它将它们转换为节点形式,实现了医疗实体之间稀疏关系的统一处理。(b) 聚合增强模块旨在聚合语义信息。成对表示集成部分利用类型信息来整合头部实体节点 \(h\) 和头部关系节点 \(r_h\),以及尾部实体节点 \(t\) 和尾部关系节点 \(r_t\)。然后,语义转移部分结合了头部和尾部信息。它采用即插即用的双向注意力机制来整合实体和关系的表现,从而获得增强的表现。(c) 交叉变换器由两层组成:特征分离层和融合层。在特征分离层中,模型分别对实体 \(X_e\) 和关系 \(X_r\) 进行编码。它们在特征融合层中被融合,并且模型通过扁平化学习全局特征。这种方法允许模型捕获丰富的实体和关系特征,同时通过特定的交叉注意力机制实现跨实体类型的语义传播。(d) GCN通过卷积操作从多跳邻居中提取有价值的信息,然后聚合局部邻域信息。
3.2 三元组变换
我们在三元组转换模块中将知识图谱三元组转换为其他多实体元组。例如,在DDI任务中,我们将三元组转换为五元组:\(\mathrm{(drug_1,r,drug_2) \rightarrow ((drug_1,r_1),r,(drug_2,r_2))}\)。如图1所示。具体来说,我们将实体的属性信息视为逻辑查询图中的关系,并将这些关系转换为关系节点,将知识图谱转换为无边属性的有向图。在实践中,三元组转换计算成本低廉,因为药物等医疗实体之间的关系图通常非常稀疏。
通过三元组转换,我们将异构图中的实体和关系信息转换为查询图中的节点。统一的节点嵌入表示使我们能够在GCN中对实体和关系执行相同的处理。
我们还添加了特定的节点类型嵌入来区分实体节点嵌入和关系节点嵌入,这对我们后续对实体和关系的独立操作有利。
3.3 表示增强模块
3.3.1 成对表示集成
整合实体、关系和节点类型的信息可以帮助在嵌入查询时平滑决策边界。但这需要捕获知识图谱中查询的目标匹配。例如,对于查询“老年人严重Covid-2019的治疗方法是什么?”,我们需要关注“治疗”在“Covid-2019”中的连接,而不是“传播”。同样,我们只应考虑从“Covid-2019”出发的“治疗”连接,而不是从“流感”或“感冒”出发的连接。为了在三元组中正确捕获这个约束,我们引入了双向注意力机制来整合每一对表示状态(“实体-关系”,“实体-类型”和“关系-类型”)。这里,实体“老年人”和关系“年龄”之间的双向整合表示可以计算为:
其中 \(\mathrm{\mathbf{h} \in \mathbb{R}^{d \times 1}}\) 是实体节点“老年人”的嵌入,\(\mathrm{\mathbf{r}_h \in \mathbb{R}^{d \times 1}}\) 是关系节点“年龄”的嵌入,\([~~]\) 是连接操作,\(\mathrm{\mathbf{W}_1,\mathbf{W}_2 \in \mathbb{R}^{2d \times 2d}}\) 和 \(\mathrm{\mathbf{b}_1,\mathbf{b}_2 \in \mathbb{R}^{2d \times 1}}\) 是可学习参数。我们采用逐元素相减 \(\ominus\) 和逐元素相乘 \(\otimes\) 来构建匹配表示。\(\mathrm{ \mathbf{M}^{er}_h \in \mathbb{R}^{2d \times 1}}\) 是将关系信息整合到实体的输出,\(\mathrm{ \mathbf{M}^{re}_h \in \mathbb{R}^{2d \times 1}}\) 是将实体信息整合到关系的输出。同样,我们可以得到将类型信息整合到实体的输出 \(\mathrm{ \mathbf{M}^{es}_h \in \mathbb{R}^{2d \times 1}}\) 和将类型信息整合到关系的输出 \(\mathrm{ \mathbf{M}^{rs} \in \mathbb{R}^{2d \times 1}}\)(类型信息是从数据集中提取的)。
通过实体和关系的双向融合,我们同时获得一个关系感知的实体表示 \(\mathrm{ \mathbf{M}^{er}_h}\) 和一个实体感知的关系表示 \(\mathrm{ \mathbf{M}^{re}}\),捕获实体及其关系互动。
接下来,我们使用门控机制来结合双向融合的结果,因为它更好地调节信息流。以实体融合表示为例,使用关系感知实体 \(\mathrm{ \mathbf{M}^{er}}\) 和类型感知实体 \(\mathrm{ \mathbf{M}^{es}_h}\) 表示作为输入,计算实体的最终表示如下:
其中 \(\mathrm{\mathbf{W}_3,~\mathbf{W}_4 \in \mathbb{R}^{2d \times 2d}}\) 和 \(\mathrm{\mathbf{b}_3,\mathbf{b}_4 \in \mathbb{R}^{2d \times 1}}\) 是可学习参数。\(\mathrm{\mathbf{G} \in \mathbb{R}^{2d \times 1}}\) 是重置门。"\(\cdot\)" 表示哈达玛德积。\(\mathrm{\widetilde{\mathbf{M}}^e \in \mathbb{R}^{2d \times 1}}\) 是最终的实体表示。
我们使用线性层将融合特征投影到原始向量大小:
其中 \(\mathrm{\mathbf{W}_5 \in \mathbb{R}^{d \times 2d}}\) 和 \(\mathrm{\mathbf{b}_5 \in \mathbb{R}^{d \times 1}}\) 是可学习参数。\(\mathrm{\mathbf{\bar{h}} \in \mathbb{R}^{d \times 1}}\) 是通过关系和类型增强的最终实体表示。
3.3.2 语义传输
成对表示集成模块主要集成实体和关系,但需要学习药物实体与药物实体之间或药物实体与相互作用之间的复杂语义。因此,我们利用一组专门的传递函数来显式地表达它们之间的语义:
在这里,\(\mathrm{\mathbf{t}}\) 是实体节点的嵌入,例如,“Covid-2019”。\(\mathrm{\mathbf{r}_h}\) 和 \(\mathrm{\mathbf{r}_t}\) 分别表示 \(\mathrm{\mathbf{h}}\) 和 \(\mathrm{\mathbf{t}}\) 的关系信息。
由于生物医学知识图中关系的数量远少于实体类型的数量,过于复杂的增强函数可能会导致关系建模中的过度拟合。为了避免这个问题,实体和关系增强函数在传递时必须具有不同的形式。
3.4 Cross-Transformer & GCN解码器
我们设计了一个两层策略,包括 Cross-Transformer 模块的药物特征分离层和融合层。在分离层中,实体和关系被分开编码。在融合层中,我们融合这两类特征并使用展平操作来学习全局特征。在GCN模块中,我们聚合了上述模块中学习到的特征,通过卷积函数学习局部特征,并进一步收集多跳邻居信息。
3.4.1 药物特征分隔层
将关系边表示为节点后,使用相同的方法分别对实体和关系节点进行编码。值得注意的是,生物信息学中关系类别的数量通常远少于实体类别的数量。因此,复杂的关系编码可能表现不佳,并且关系编码器的层数通常比实体编码器少。在这里,我们演示如何对实体节点进行编码。
给定第 \(k\) 个药物中实体节点 \(u\) 的嵌入 \(\mathrm{\mathbf{x}_u^{(0)}} \in \mathbb{R}^\mathrm{d}\)特征分离层,\(\mathrm{d_h=\frac{d}{h}}\),其中\(\mathrm{h}\)表示注意力头的数量。多头注意力的计算公式为:
其中\(\mathrm{\mathbf{Q}=\mathbf{x}}^{(k-1)}_\mathrm{u}\mathrm{\mathbf{W}_Q}\)和\(\mathrm{\{\mathbf{K},\mathbf{V}\}={\|}_{n \in \mathcal{N}(u)}} \mathrm{\mathbf{x}}^{(k-1)}_\mathrm{n} \{ \mathrm{\mathbf{W}_K,\mathbf{W}_V}\}, \mathrm{\mathbf{W}_{\{Q,K,V\}}\in} \mathrm{\mathbb{R}^{d \times d_h}}\),\(\|\)表示串联,\(\mathrm{\mathcal{N}_u}\)指节点\(的邻居集合\mathrm{\mathcal{N}(u)}\)。
接下来,我们在由\(\mathbf{W}_{\mathrm{O}} \in \mathbb{R}^{d \times d}\)投影的注意力权重的输出上应用前馈神经网络:
其中 \(\mathrm{\mathbf{W}_6, \mathbf{W}_7 \in \mathbb{R}^{d \times d}}\) 和 \(\mathrm{\mathbf{b}_6,~\mathbf{ b}_7 \in \mathbb{R}^{d}}\) 是可学习的参数。 FNN 不会改变矩阵大小,但可用于捕获数据中的模式。 FNN 相当于一个基于记忆和注意力的模型的键值网络。
3.4.2 药物特征融合层
如图1所示,在获得药物特征分离层的输出之后,我们使用两个融合编码器来聚合实体和关系,然后通过扁平化和全连接层学习药物的全局特征。这里,我们展示了如何聚合实体和关系:
给定药物特征分离层实体编码输出嵌入 \(\mathrm{\mathbf{x}^{(0)}_e \in \mathbb{R}^d}\) 和药物特征分离层关系编码输出 \(\mathrm{\mathbf{x}^{(0)}_r \in \mathbb{R}^d}\),在第 \(k\) 个药物特征分离层中,\(\mathrm{d_h}=\frac{d}{h}\),其中 \(\mathrm{\mathbf{h}}\) 表示注意力头的数量。多头注意力的计算公式为:
其中 \(\mathrm{\mathbf{Q=x}}_e^{\mathrm{(k-1)}}\mathrm{\mathbf{W}_Q,\{\mathbf{K,V}\}=\mathbf{x}_r^{(k-1)}}, \mathrm{\mathbf{\mathbf{W}}_{\{Q,K,V\}}} \in \mathbb{R}^{d \times d_h}\)。接下来,与药物特征分离层一样,我们在通过 \(\mathrm{\mathbf{W}_O \in \mathbb{R}^{d \times d}}\) 投影的注意力权重上应用前馈神经网络 \(\mathrm{FFN(x)}\):
其中 \(\mathrm{\mathbf{W}_8,\mathbf{W}_9 \in \mathbb{R}^{d \times d}}\) 和 \(\mathrm{\mathbf{b}_8,\mathbf{b}_9 \in \mathbb{R}^d}\) 是可学习的参数。与药物特征分离层不同,两个融合编码器的层数应该相等,因为实体和关系在融合层中被融合和聚合,并且地位相等。最后,融合输出 \(\mathrm{\mathbf{x}_e}\) 和 \(\mathrm{\mathbf{x}_r}\) 被扁平化以学习全局特征:
其中 \(\mathrm{\mathbf{W}_{10} \in \mathbb{R}^{2d \times d}}\), \(\mathrm{\mathbf{W}_{11} \in \mathbb{R}^{d \times d}}\) 和 \(\mathrm{\mathbf{b}_{10}, \mathbf{b}_{11} \in \mathbb{R}^d}\) 是可学习的参数,\(\mathrm{\mathcal{H}\in \mathbb{R}^{d}}\) 是融合层的最终输出。
3.4.3 GCN解码器
在TransFOL模型中,我们采用图卷积网络作为解码器组件,其主要目的是学习并聚合药物实体及其邻域节点的局部特征以完成药物相互作用预测任务。
具体来说,在之前融合层模块学习全局特征的基础上,GCN通过在知识图谱上进行信息传播和聚合操作,能够捕捉到每个药物节点与其邻居节点之间的内在联系和相似性。这些局部特征不仅包含药物自身的属性信息,还包含了与之相连的其他生物医学实体(如疾病、靶点等)的相关知识。
除了提取局部特征,每一层GCN都会递归地将当前节点的信息与其一跳或多跳邻域的信息结合,形成一个更丰富、更全面的节点表示。这种多层级的邻域融合使得模型能够从不同层次理解和挖掘潜在的药物交互关系。
在经过Cross-Transformer编码获得药物及关系的高级语义表示之后,GCN模块负责收集和整合这些上下文信息,用于推断出最终的药物相互作用类别或强度,增强了模型对复杂情境下DDI预测的准确性和泛化能力。
单层图卷积的聚合方法如下所示:
其中 \(\mathcal{H}^{(k+1)}_i \in \mathbb{R}^{d^{(k)}}\) 表示在 GCN 第 \(k\) 层中节点 \(i\) 的隐藏状态,\(\mathrm{d}^{(k)}\) 表示第 \(k\) 层节点嵌入的维度,\(\mathrm{\mathbf{W}}_{\tau}^{(k)}\) 是连接第 \(k\) 层与节点 \(i\) 的边的权重。\(\mathcal{N}^i_{\tau}\) 表示节点 \(i\) 的直接邻居节点集,包括节点 \(i\) 本身。
3.5 训练和推理
在这项研究中,我们利用标记的监督信息来训练模型。具体来说,我们使用推理数据集来训练模型,该数据集包含遮蔽预测。数据集包含一组遮蔽查询图的可能答案。给定一组可行答案 \(\mathcal{E}_{\mathrm{A}}\) 以及遮蔽查询图的可训练输入嵌入 \(\mathbf{x}\),每个查询的损失函数定义为:
其中 \(\mathrm{\mathbf{u}_e}\) 指的是解码器中实体 \(\mathrm{e}\) 的嵌入。在计算一个答案的损失时,其他答案的预测在损失函数中被忽略。
在训练期间,模型通过使用随机梯度优化器AdamW来最小化总损失来学习。这种方法使模型能够根据标记的监督信息优化其参数。
在推理过程中,提取知识图中未知节点对 \(\mathrm{(u, v)}\) 的子图。为了预测 \(\mathrm{u}\) 和 \(\mathrm{v}\) 之间的关系,我们使用与训练过程中相同的步骤来计算关系向量。在多分类任务中,选择最高概率的关系作为预测关系。相比之下,对于多标签任务,收集所有关系的正面和负面对应物的所有分数,以预测关系。这种方法允许模型泛化到新实例并进行准确预测。
4.实验
这一节对TransFOL在四个药物推理任务中的逻辑查询能力进行了全面评估。为了评估模型的性能,我们使用表1中描述的四个不同的数据集进行了四项任务。
4.1 任务描述
这一节详细介绍了四个任务。四项任务的逻辑查询过程如表1所示。
| Task | Logical Query Process |
|---|---|
| 1 | \(\{((drug_1, drug_2))\} \rightarrow \{interaction\}\) |
| 2 | \(\{((drug_1, drug_2))\} \rightarrow \{interactions\}\) |
| 3 | \(\{((drug_1,r_1),(drug_2,r_2))|r_1,r_2 \in doses\} \rightarrow \{response\}\) |
| 4 | \(\mathrm{train:\{(MP_i},r_{m1},r_{m2})\} \rightarrow \{\mathrm{HP_j}\},\{(disease,r_{d2})\} \rightarrow \{\mathrm{HP_j}\}\) \(\mathrm{test:\{((MP_i},r_1,r_2),(disease,r_3))\} \rightarrow \{\mathrm{HP_j}\}\) |
表1:四个任务的具体逻辑查询流程。
在任务1和任务2中,我们专注于常见的药物相互作用(DDI)任务。任务1评估模型预测一对药物之间唯一的DDI类型的能力。任务2评估模型预测多种共现DDI类型的能力。
不同药物组合的不同剂量可能对目标细胞产生不同的影响。因此,在任务3中,我们进行多剂量药物组合-目标反应预测。在这个任务中,预测的锚点节点不仅限于简单的药物对,而是由不同剂量的药物对组成。这样的带有属性信息的预测更加可靠和现实,对现有的预测方法来说是一个挑战,这表明TransFOL在重要的药物知识发现能力和更好地捕获丰富知识信息方面比现有的DDI模型具有更强的能力。在任务3中,我们评估模型预测不同剂量下多种药物组合对目标的影响的能力,这对于缩短药物开发时间至关重要。
此外,在任务4中,我们模拟了一个药物研究实验的临床阶段,仅基于疾病信息和哺乳动物表型(MP)推断人类临床表型(HP)。我们的方法学习了Phenomebrowser异构图,并仅输入疾病和MP信息。需要指出的是,最终预测的HP信息与数据集中的MP信息没有直接关系,通常相关的药物只决定了它们的关系。因此,在测试期间,模型只能依靠其对异构图的理解以及泛化能力来进行预测。
我们研究的目标是基于哺乳动物的反应和相应的疾病信息来预测未知药物对人类的临床反应。在测试阶段,当输入实体对(“哺乳动物症状,疾病信息”)时,模型可以预测人类的症状数据。训练集不包括实体-关系对,以评估模型的泛化能力和可转移性。
任务4包括药物知识发现任务,如药物-蛋白质相互作用(DPI)、药物目标相互作用(DTI)和药物-人类相互作用(DHI)。现有的药物知识发现模型无法解决这样复杂的任务,突显了TransFOL的独特优势。现有的药物知识发现模型无法解决这样复杂的任务。同时,TransFOL可以通过查询转换来适应不同类型的实体关系,实现语义学习,这充分展示了TransFOL的优势。
4.2 基线方法
我们将我们的模型与几个基线模型进行比较。所有基线模型的参数设置都是原始文本中的最优参数。由于现有的DDI模型在处理任务3和任务4方面存在困难,因此仅选择了逻辑查询基线进行比较。
- GQE使用深度集合进行交集(合取)逻辑操作,作为在此空间中学习的几何函数。
- GraIL用于知识图谱上的归纳关系预测,它使用局部子图。
- KGNN采用GCN选择性地聚合带有高层级信息的邻居信息,以学习知识图谱中的节点表示。
- Q2B将逻辑查询和知识图谱实体嵌入到几何向量空间中,以箱形表示。
- Logic利用复杂查询嵌入,并使用Skolem化来消除存在变量,以实现高效查询。
- ConE是一种基于几何的查询嵌入(QE)方法,将实体和查询表示为二维锥体的笛卡尔积,并且可以处理所有FOL操作。
- MUFFIN采用双层交叉策略,包括交叉层和标量层组件,以很好地融合多模态特征。
- SumGNN采用子图提取模块和基于自注意力的子图摘要方案来预测DDI。
4.3 实验设置
4.3.1 数据集
在这项研究中,我们使用四个不同的数据集来执行四项任务。
(i) 对于多类DDI预测任务,我们使用DrugBank数据集,它包含1709种药物(节点)和136,351对药物对(边),有86种药理关系。每对药物可能包含一个或两个关系。由于超过99.8%的边只有一种边类型,我们在研究中过滤掉了有多种类型的边。
(ii) 对于多标签DDI预测任务,我们使用TWOSIDES数据集,它包含645种药物(节点)和46,221对药物-药物对(边),有200种不同的药物副作用类型作为标签。每条边可能与多个标签相关联。我们保留了200种常见的DDI类型,范围从Top-600到Top-800,以确保每种DDI类型至少有900种药物组合。
(iii) 对于多剂量药物组合-目标反应预测任务,我们使用DrugCombDB数据集,它基于SPARQL查询,目前包含超过400万种基因、疾病、药物、病原体和化学实体(代谢物)的表型关联和6,055,926个实验数据点,其中包含有关癌症细胞的药物剂量-反应和药物浓度信息,涵盖2,887种抗癌药物和104种癌症细胞类型。
我们从中提取了一个包含“\textit{药物-剂量-目标-反应}”关系的子图数据集。
这个子数据集介于传统的DDI数据集和大规模生物医学数据集之间,整合了药物不同剂量条件下与其在目标上的生物学效应之间复杂的交互信息。
就社区使用而言,这个子数据集适用于预测不同剂量下的药物组合-目标反应,并支持各种药物发现和药理学研究任务。这些任务包括研究药物剂量变化对药物疗效和副作用的影响,优化药物组合治疗方案,解读药物的作用机制。此外,研究人员可以利用这个数据集进行二次分析,结合机器学习方法来揭示潜在的剂量-反应模式,从而有助于减少药物开发周期并提高治疗效果。
(iv) 对于模拟临床试验任务,我们使用Phenomebrowser数据集,它包含从多个数据源提取的交互信息,总共包含61,146个医疗实体(节点),1,246,726对医疗实体对(边),七种节点类型和九种语义关系。每种生物医学概念(即药物、基因、疾病和表型)被视为一种节点类型,每种交互(即药物-靶标-基因、药物-治疗-疾病)被视为一种边类型。我们从中提取包含“\textit{哺乳动物症状-人类症状-药物-疾病}”关系的子数据集。
子数据集展示了各种结构化连接,如药物-靶标相互作用(药物-靶标-基因)的分子机制和药物在实际治疗中的治疗效果(药物-治疗-疾病)等复杂关系。它的目的是在临床环境中模拟症状反应和药物疗效评估,为跨物种相关性分析提供独特的视角。这个子数据集不仅适用于模拟临床试验预测任务,也适用于探索症状的演变路径,验证药物指示的有效性和安全性,揭示药物效应的潜在生物学机制,以及通过比较不同物种的症状表现来揭示疾病的共性和差异,在生物医学研究场景中揭示疾病的共性和差异。
4.3.2 评估策略
我们将四个数据集按照7:1:2的比例划分为训练集、验证集和测试集。为确保高度不平衡的标签分布不影响模型的性能,我们确保所有数据集的训练/验证/测试集之间没有重叠,保持它们的独立性。
4.3.3 度量指标
我们使用不同的指标来衡量TransFOL在四个不同任务中的性能。此外,我们还设计了一个指标 \(\mathrm{SUB=|Prediction-Truth|}\) 来衡量任务3中预测值和实际值之间的平均差异。当数据精度高时,指标 \(\mathrm{SUB}\) 可以更真实地评估预测结果接近但只有部分一致的情况。
4.3.4 参数设置
我们将知识图谱实体、关系和药物结构嵌入表示为1024维的向量。使用AdamW优化器来优化训练损失。实验学习率设置为0.0001。模型训练了500个周期。基线模型的超参数设置与它们原始论文中报告的相同。在没有额外信息的任务1和任务2中,分离层中的实体编码器层数和关系编码器层数分别为2和1;而在有复杂查询的任务3和任务4中,两者都是两层,这由任务数据集中关系类型的数目决定。
融合层的编码器层数为八层。最后一个全连接层的隐藏层大小设置为2048。参数敏感性实验发现,在任务1和任务2中,最佳的GCN解码器层数是3,在任务3和任务4中,最佳层数是1。完整的和详细的参数设置信息请见附录III。
4.4 实验结果
4.4.1 多分类DDI预测
我们在表2中展示了我们的模型的结果以及 Drugbank 数据集上的多类 DDI 预测任务的所有基线。我们的模型在预测 DDI 方面优于所有基线。具体来说,与最佳基线相比,我们的模型分别实现了 0.89%(Accuracy)、0.13%(F1-Score)和 5.46%(Cohen's Kappa)改进。
| Methods | Multi-class | Multi-class | Multi-class | Multi-label | Multi-label | |
|---|---|---|---|---|---|---|
| Accuracy | F1-Score | Cohen's Kappa | AUC | AUPR | ||
| GraIL | 0.8989 | 0.8131 | 0.8989 | 0.9289 | 0.9110 | |
| KGNN | 0.9089 | 0.8291 | 0.8964 | 0.9075 | 0.8816 | |
| MUFFIN | 0.9648 | 0.9495 | / | 0.9160 | 0.7033 | |
| SumGNN | 0.9266 | 0.8685 | 0.9072 | 0.9486 | 0.9335 | |
| TransFOL | 0.9737 | 0.9508 | 0.9618 | 0.9487 | 0.9426 |
表2:多类和多标签 DDI 预测任务的结果。
Note: 粗体字体的方法表示最佳方法,带下划线字体的方法表示次优方法。较高的值始终表示这些指标的性能较好(SUB 除外)。以下同样适用。
4.4.2 多标签DDI预测
我们在表2中展示了我们的模型的结果以及 TWOSIDES 数据集上多标签 DDI 预测任务的所有基线。 TransFOL 在多标签任务中表现最好。具体来说,与最佳基线相比,它分别实现了 0.01% (AUC)、0.91% (AUPR) 和 1.54% (AP@50) 的改进。
4.4.3 多剂量药物联合靶标反应预测
| Methods | Hits@1 | Hits@3 | Hits@10 | F1-Score | SUB |
|---|---|---|---|---|---|
| GQE | 0.7663 | 0.8593 | 0.9261 | 0.6949 | 9.7440 |
| Q2B | 0.7399 | 0.8310 | 0.9017 | 0.7002 | 12.5288 |
| LogicE | 0.7507 | 0.8787 | 0.9652 | 0.6943 | 9.5574 |
| ConE | 0.7466 | 0.8324 | 0.9277 | 0.7066 | 11.2375 |
| TransFOL | 0.7760 | 0.9090 | 0.9776 | 0.7101 | 9.0998 |
表3:多剂量药物组合反应预测任务的结果。
Note: 由于任务3中的实体具有多个属性信息,现有的DDI模型无法完成,因此所有基准模型都是逻辑查询模型,任务4也是如此。
我们使用表3中的 DrugCombDB 数据集展示了模型的性能以及多剂量药物组合反应预测任务的所有基线。结果表明 TransFOL 在复杂环境中实现了最佳性能。具体来说,与最佳基线相比,我们的模型分别实现了 0.97% (Hits@1)、3.03% (Hits@3)、1.24% (Hits@10)、0.35%(F1-Score) 和 0.4576%(SUB) 的改进。
4.4.4 模拟临床试验
| Methods | Hits@1 | Hits@3 | Hits@10 | F1-Score |
|---|---|---|---|---|
| GQE | 0.1518 | 0.2867 | 0.3386 | 0.5528 |
| Q2B | 0.1727 | 0.2733 | 0.4360 | 0.5257 |
| LogicE | 0.1727 | 0.2733 | 0.4360 | 0.4833 |
| ConE | 0.1521 | 0.2780 | 0.4240 | 0.5444 |
| TransFOL | 0.1881 | 0.3027 | 0.4744 | 0.5554 |
表4:模拟临床试验结果。
表4显示了我们的模型的性能和使用Phenomebrowser数据集的模拟临床试验中的所有基线。结果表明,我们的模型 TransFOL 实现了最高性能。具体来说,与最佳基线相比,我们的模型分别实现了 1.54% (Hits@1)、1.60% (Hits@3)、3.84% (Hits@10) 和 0.26%(F1-Score) 的改进。
4.5 消融实验
| Modules | Multi-class | Multi-class | Multi-label | Multi-label | Multi-dose | Multi-dose | Simulated | Simulated | Simulated | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy | F1 | AUC | AUPR | Accuracy | SUB | Hits@1 | Hits@3 | Hits@10 | ||||
| w/o EN & GCN | 0.9563 | 0.9052 | 0.8678 | 0.7939 | 0.7657 | 9.2305 | 0.1713 | 0.2405 | 0.3632 | |||
| w/o EN | 0.9673 | 0.9136 | 0.9383 | 0.9285 | 0.7742 | 9.1024 | 0.1808 | 0.2943 | 0.4632 | |||
| w/o GCN | 0.9658 | 0.9105 | 0.8820 | 0.8220 | 0.7673 | 9.2213 | 0.1789 | 0.2538 | 0.3855 | |||
| TransFOL | 0.9737 | 0.9508 | 0.9487 | 0.9426 | 0.7760 | 9.0998 | 0.1881 | 0.3027 | 0.4744 |
表5:四项任务的消融实验结果。 EN - Enhancement.
增强模块与GCN。 我们对四个实验进行了消融研究,以分析增强模块和GCN的功能。表5报告了所有任务中不同模型结构预测的平均结果。没有增强模块,模型在四个数据集上的性能指标显著下降,分别为0.64%(Accuracy)、1.04%(AUC)、0.18%(Accuracy)和0.73%(Hits@1)。同样,没有GCN解码器,模型在四个数据集上的性能指标分别下降了大约1.15%(Accuracy)、6.67%(AUC)、0.87%(Accuracy)和0.92%(Hits@1)。此外,如果没有增强模块和GCN解码器,模型在四个数据集上的性能指标下降得更多,这表明增强模块和GCN都可以提高模型对复杂查询推理的能力。需要指出的是,分离层和融合层是模型的基础,因此我们没有对它们进行消融实验。
超参数。 我们对GCN层数进行了消融实验,并在图2中报告了每次实验的结果。在任务1和任务2中,随着GCN层数的增加,模型的性能先是提高然后降低,这表明拥有足够和适当的模型深度对于TransFOL的成功至关重要。深度和接受场大小之间的关系在先前的研究中已经被注意到,表明过深的GNN可能会受到过平滑和训练不稳定的影响,导致性能下降。然而,在任务3和任务4中,随着GCN网络层数的增加,模型的性能逐渐下降。此外,消融实验表明,没有GCN解码器,模型的性能更差。我们认为,这可能是由于任务3和任务4的数据集规模相对较小,使它们更容易过拟合。
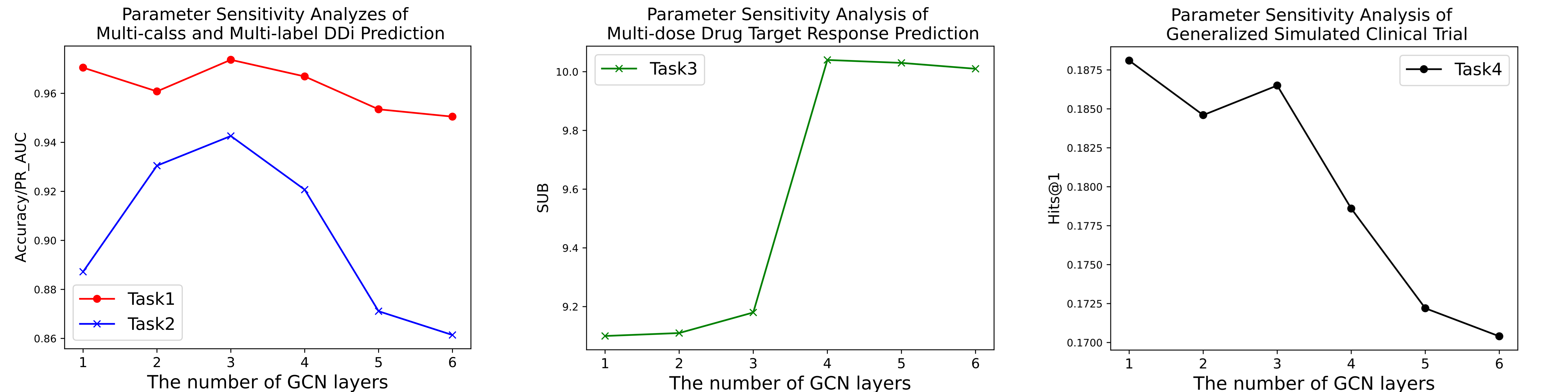
图2:参数敏感性分析
4.6 案例研究
| 药品名称 | 互动类型 | 证据 |
|---|---|---|
| 伊利鲁司他 | 与奎尼丁合用时,Eliglustat的代谢会降低。 | Drug.com |
| 碳酸氢钾 | 碳酸氢钾可能会降低奎尼丁的排泄率,从而导致血清水平升高。 | PMID: 5360313 8739822 |
| 柳氮磺吡啶 | 柳氮磺吡啶可能会降低奎尼丁的排泄率,从而导致血清水平升高。 | DrugBank |
| 长春新碱 | 长春新碱是一种长春花生物碱,用于治疗急性白血病、恶性淋巴瘤、霍奇金病、急性红斑病和急性全骨髓病。 | DrugBank |
| 布雷蒂鎓 | 布雷蒂鎓可能会增加奎尼丁的降血压活性。 | PMID:386786 |
| 钾 | 钾可能会降低奎尼丁的排泄率,从而导致血清水平升高。 | PMID: 8930193 6025406 |
| 赖诺普利 | 赖诺普利是一种血管紧张素转换酶抑制剂,用于治疗高血压、心力衰竭和急性心肌梗塞。 | 未经确认 |
| 碘丘氨酸 | 碘丘氨酸可能会降低奎尼丁的排泄率,从而导致血清水平升高。 | DrugBank |
| 罗米地辛 | 当罗米地辛与奎尼丁合用时,QTe 延长的风险或严重程度可能会增加。 | PMID:32449208 |
| 环孢素 | 环孢素是一种节省类固醇的免疫抑制剂,用于器官和骨髓移植以及炎症性疾病。 | DrugBank |
表6:可能与奎尼丁相互作用的 10 种药物。
这一节通过案例研究展示了TransFOL的预测能力。
首先,在多类DDI任务中,我们对TransFOL的预测结果进行了详细分析。按照Yu等人的研究,我们关注了奎尼丁(Quinidine)的前10个药物对,如表6所示。我们使用了PubMed、DrugBank和Drugs.com提供的药物相互作用检查工具来验证这十个药物对的证据。表6显示,10个DDI对中有9个有支持证据,进一步证明了TransFOL的有效性。例如,我们的模型预测Eliglustat与奎尼丁合用时会减少代谢,这在文献中已有报道。此外,模型预测的药物对(洛普利(Lisinopril),奎尼丁)并未包含在数据集中。这可能是一个潜在的DDI对,Shulyak等人对此进行了一些研究,确认了洛普利和奎尼丁之间的关联。
其次,在多标签DDI任务中,我们提供了模型预测的新药对——氯雷他定(loratadine)和昂丹司琼(ondansetron)的案例研究。氯雷他定是一种过敏药物,而昂丹司琼用于预防或治疗化疗后的恶心和呕吐。TransFOL对视力丧失、运动迟缓和乳腺疾病的三个副作用节点赋予了高权重。视力丧失是感知视觉刺激能力下降;运动迟缓是一种患者基本反射和运动受限的疾病状况。此外,氯雷他定和昂丹司琼的组合可能导致影响乳腺的一些非肿瘤或肿瘤状况。值得注意的是,视力丧失与运动迟缓密切相关,这是由抑郁情绪引发的中枢神经系统问题。由于两种药物都有副作用风险,我们的模型预测它们一起使用可能会导致中枢神经系统(CNS)恶化,这在文献中得到了支持。
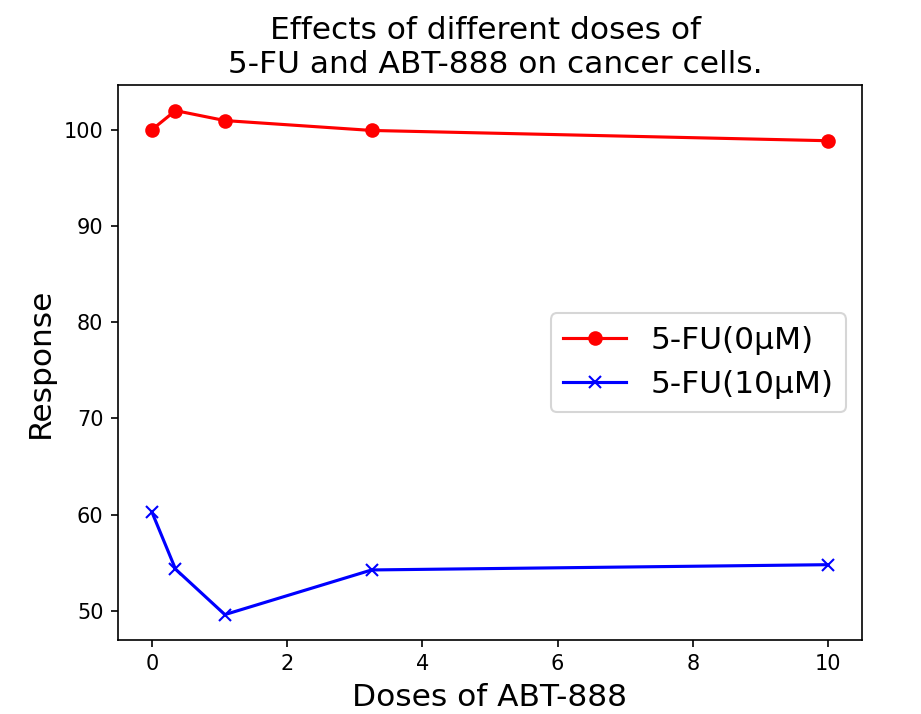
图3:不同剂量的 5-FU 和 ABT-888 对癌细胞的反应。 注:反应是指癌细胞的活性;值越低代表药物的抑制效果越好。
第三,在多剂量药物组合-目标反应预测任务中,我们还提供了模型预测的不同剂量组合对5-FU和ABT-888的案例研究。5-FU是一种胸苷酸合成酶抑制剂,广泛用于癌症治疗,而ABT-888(Veliparib)是一种PARP抑制剂。如图3所示,根据模型预测,当5-FU的剂量为0 \(\mathrm{\mu M}\)时,癌细胞的反应值一致较高(值越低越好)。然而,当5-FU的剂量为10 \(\mathrm{\mu M}\)时,随着ABT-888剂量的增加,癌细胞的反应值降低。总体水平较低,意味着单独的ABT-888几乎没有抗癌效果。在一定剂量的5-FU之后,随着ABT-888的添加,抗癌效果增加。这一结果已在文献中报告。
最后,在模拟的临床试验中,我们提供了模型预测的新药临床试验的案例研究。据报道,为脓疱病开发的一种特定新药可以在哺乳动物实验模型中引起生理稳态的失调。根据模型预测,在人类临床试验中可能会出现皮肤光敏感性和唇炎等不良反应,其中皮肤光敏感性被赋予最高的权重,这已在文献中报告,并进一步证明了TransFOL的有效性,并证明了TransFOL可以帮助缩短药物开发的临床试验周期。
4.7 模型局限
虽然 TransFOL 在复杂场景中表现出了良好的推理能力,但也存在一些局限性:
错误累积. 生物医学数据集是稀疏且存在大量噪声的,因此在多跳推理过程中,TransFOL可能学习到错误知识并随着跳数增加而将错误推广。例如,如果输入的训练数据包含错误的信息“药物A与药物B相互作用”,那么在推理阶段,我们的模型将进一步推断出“与药物A属于同一类型且具有相似的邻居关系的药物C与药物B相互作用”,然后又推断出“与药物B相似的药物D与药物C相互作用”。。。然而,这些DDI关系并不存在。然而,这些关系并不存在。未来研究中我们将尝试引入基于类型的负采样策略和对抗学习等技术来帮助模型更好地纠正错误。
查询模板化. 虽然TransFOL能够进行多种复杂预测,但这都需要将数据预处理成对应的固定格式,这种模板化的查询方式限制了模型的应用。例如,为满足固定的查询模板,可能需要额外的人工干预和数据预处理工作,增加了从原始数据到模型可接受格式转化的成本,并可能导致潜在有用信息的损失;同时,随着知识库的增长和更新,新的发现和数据可能不符合现有查询模板,使得模型难以快速吸收和利用这些新知识进行实时更新和预测。未来研究中我们将尝试采用图结构化查询来替代固定的数据模板,以及通过学习数据的特征和模式,动态生成适应性查询。
图结构化查询的关键优势在于其能够以更灵活的方式表示和查询药物-药物相互作用。与固定数据模板相比,图结构化查询可以根据特定数据特征和模式动态生成查询语句,更好地适应不同场景。然而,尽管图结构化查询在理论上具有吸引力且潜在有益,我们在当前研究中并未直接采用这种方法,因为需要进行更深入的研究来充分解决使用图结构化查询的挑战。采用图结构化查询可能需要重新设计和重新训练模型,以更好地适应新的查询方法,这可能涉及更复杂的模型架构和算法,以及额外的数据和计算资源来支持模型的训练和推理。
结论
本文提出了一种用于复杂 DDI 预测的新方法 TransFOL。我们基于逻辑查询形式并使用Cross-Transformer模块对实体和 GCN 解码器之间的复杂语义信息进行编码,以进行进一步的解码集成。 我们在生物医学数据集上的实验表明,TransFOL在跨多个任务的四个基准数据集上实现卓越的性能,并超越多个强大的基线,展示了它强大的药物知识发现能力和泛化能力。
附录
I.逻辑查询模式
如图4所示,实验中常用的逻辑查询有14种类型。我们在任务 1、任务 2 和任务 3 中使用“\(2i\)”类型查询类型进行推理。对于任务 4,我们在任务 4 中使用“\(1p\)”类型和“\(2p\)”类型查询类型训练阶段,最后使用“\(pi\)”类型的查询进行推理,以评估模型的泛化能力。
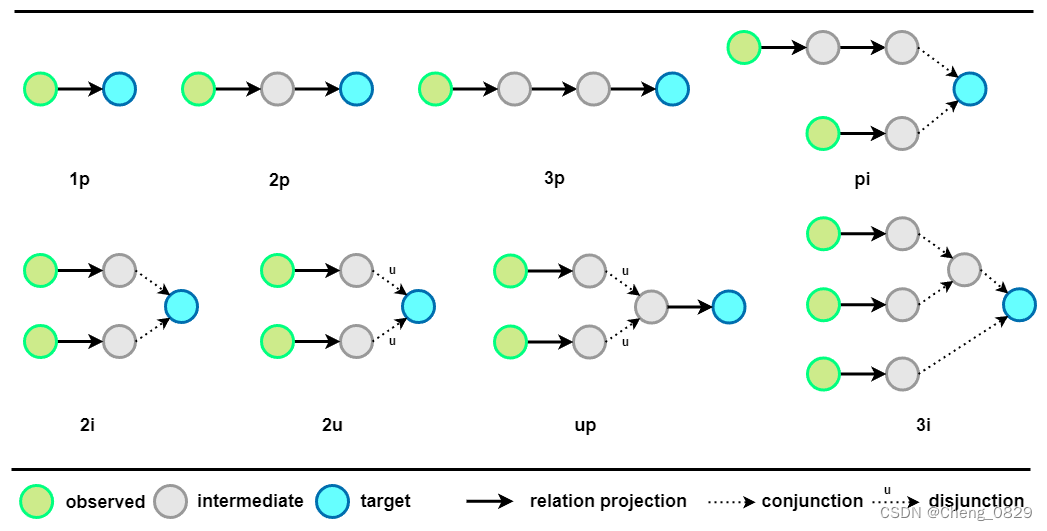
图4:常见逻辑查询模式
II.析取范式分解
本节举例说明如何将析取范式分解为多个合取范式:
III.参数设置
标签平滑. 标签平滑参数通常用于减少标签的过度自信,增强模型的泛化能力和鲁棒性。在本研究中,通过在训练过程中对真实标签应用KL散度平滑来实现标签平滑。在任务1和任务2中,由于训练集规模较大,不需要标签平滑,标签平滑参数为0;而在任务3和任务4中,数据量较小,因此需要将标签平滑参数设置为0.1以防止过拟合。
编码器层. 在实验中,我们将编码器分为两部分——药物特征分离层和融合层。在分离层中,实体和关系分别进行编码。在任务1和任务2中,没有额外的属性信息。因此,关系编码器只有一层,仅用于保存和传递关系节点嵌入,而实体编码器的层数多于任务3和任务4中的层数。相比之下,由于任务3和任务4中信息更丰富,分离层中的实体编码器和关系编码器层数设置为相同的2层。在融合层中,实体和关系同时嵌入到对方的多头注意力中,实现“融合”的目的。因此,两者的地位是相等的,在融合层中实体编码器和关系编码器的层数也是相等的;两者都是八层。
GCN解码器层. 在消融研究中,我们研究了每个任务中GCN层数对最终指标的影响。在图2中,可以观察到在任务1和任务2中,随着GCN层数的增加,模型的性能先是提高然后降低。当层数为3时,指标达到最高。然而,在任务3和任务4中,随着GCN网络层数的增加,模型的性能逐渐下降,当层数为1时,指标达到最高。通过分析,我们得出结论,这种方法是由于任务3和任务4的数据集规模相对较小。多层GCN主要用于提取局部特征和连接多跳邻域,这更容易发生过拟合。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号