知识图谱-生物信息学-医学顶刊论文(Bioinformatics-2021)-SumGNN:通过有效的KG聚集进行多类型DDI预测
3.(2021.3.26)Bioinformatics-SumGNN:通过有效的KG聚集进行多类型DDI预测
论文标题: SumGNN: multi-typed drug interaction prediction via efficient knowledge graph summarization
论文地址: https://academic.oup.com/bioinformatics/article-pdf/37/18/2988/40471587/btab207.pdf
论文期刊: Bioinformatics 2021
摘要
动机:
由于DDI数据集和大型生物医学KG的日益可用性,使用机器学习模型对不良DDI进行准确检测成为可能。然而,如何有效利用大规模但含有噪声的生物医学KG进行DDI检测仍是一个有待解决的问题。由于KG的规模和噪声都很大,直接将KG与其他更小但质量更高的数据(如实验数据)集成通常不太有益。大多数现有的方法完全忽略了KG。有些人试图通过GNN直接将KG与其他数据集成,但效果不太理想。此外,以往的研究多集中于二分类DDI的预测(例如两种药物之间有没有DDI存在),而多类型DDI的药效作用预测则更有意义且难度更大。
结果:
为了填补这一空白,我们提出了一种新的方法SumGNN:知识聚集图神经网络。它由以下模块组成:
- 子图提取模块,提取药物对周围KG的局部子图,以获得有用的信息。
- 基于自注意力的子图聚集方案,生成子图内的浓缩推理路径(即计算子图中任意一对相邻节点的边的信号强度分数,并且只保留一部分重要边)。
- 多通道知识和数据集成模块(最终药物表示=知识聚集(由每一层的信号强度分数和相邻节点进行加权求和得到节点的关系感知消息,集成到同一层实体表示)+子图均值+化学指纹)来预测药效作用。**
SumGNN的性能比最佳模型高出5.54%,在低数据关系类型中性能提高尤其显著。此外,SumGNN通过生成推理路径,可以提供可解释的预测。
1.引言
不良的药物-药物相互作用(DDI)是一种药物与另一种药物一起使用时对药物效果的改变,对于病情复杂的患者来说,这是一种常见且危险的情况。
未发现的不良DDI已成为严重的健康威胁,仅在美国每年就导致近74,000人次在急诊室就诊和195,000人次住院。为了降低这些风险和成本,准确预测DDI成为一项重要的临床任务。正在利用两种类型的数据来开发DDI检测模型:人工管理的DDI网络和大型生物医学KG。
1.1 人工DDI网络
研究人员根据实验数据集和文献人工创建DDI网络,如TWOSIDEA、MINER和DrugBank。这些经过精选的数据质量较高,但创建成本较高,而且通常较小。
1.2 KG
多年来,通过文献挖掘和数据库整合,开源了许多大型KG,例如Hetionet和DRKG。然而,这些KG规模和噪声都很大:在它们数以万计的具有数百万条边的节点中,只有一个小的子图与预测目标相关。
1.3 深度学习
GNN通过将DDI预测转换为DDI图上的链接预测问题,取得了很好的性能。然而,现有的深度学习模型往往只基于DDI数据集本身进行训练,而忽略了大型生物医学KG(因为DDI是由复杂的生物医学机制驱动的,所以有助于DDI预测)。最近的一些工作试图通过直接集成标准KG和GNN方法将KG整合到DDI预测中。但DDI预测存在单独的建模困难,因为输入的KG规模大且有噪声,而药物对的相关信息是局部的。此外,尽管预测特定的DDI类型是一项更有意义的任务,但大多数现有的工作也只进行二进制分类,即预测DDI的存在。
1.4 我们的方法
在这项工作中,我们提出了一种新的方法SumGNN,它有效地使用KG来辅助药物相互作用预测。SumGNN具有更好的预测性能、效率、归纳性和可解释性。SumGNN提供了以下技术贡献:
- 用于识别有用信息的局部子图。我们在药物对周围的KG中使用局部子图而不是整个KG来提取有用的信息。子图公式允许通过锚定相关信息来降低噪声,并且由于消息传递接受范围显著减小,因此具有高度可伸缩性。
- 用于推理路径生成的子图聚集方案。然后,我们提出了一个聚集方案来生成DDI的推理路径。我们提出了一种与层无关的自我注意力机制,为子图中的每条边产生信号强度分数,并选出分数高的KG子图路径。由于选出的子图是稀疏的,它提供了对驱动药物相互作用的生物过程的洞察。
- 多通道数据和知识集成,以改进多类型DDI预测。我们建议使用多通道神经编码来聚合不同的数据源集合,从聚集子图嵌入到化学结构。它能够利用大量的外部生物医学知识来显著改进多类型DDI预测。此外,神经编码在每次传播中采用不同的子图,形成了一种归纳偏差,从而促进了低资源DDI类型的泛化。
我们进行了大量的实验,表明SumGNN显著改善了DDI预测。它比F1的最佳基线提高了5.54%,同时大大减少了推理时间。此外,SumGNN在低资源环境下表现出色,而以前的工作并非如此。SumGNN还能够为药物相互作用的潜在机制提供合理的线索。
2.相关工作
2.1 外部KG整合
最近,有几项努力试图利用KG进行下游任务,如推荐、信息提取和DDI预测。
对于DDI的预测,有人在DDI预测时集成了药物(代谢)动力学(PK)或药效学(PD)副作用开发了一个贝叶斯网络,将分子相似性和药物副作用相似性结合起来预测药物效果。然而,这些方法只将副作用作为外部知识来考虑,这在我们的任务中可能不够全面。
随着生物医学KG的出现,更多类型的实体和关系被应用于这项任务,因为有人使用KGE技术将每个实体和关系投影到密集向量,然后将它们输入到神经网络进行预测。然而,它们在推理过程中没有直接利用目标实体的邻域信息,从而没有充分利用外部知识信息。
为了克服上述缺点,有人采用带邻域采样的图卷积网络(GCN)来显式地建模邻域关系,具有更快的推理速度。然而,由于每个相邻实体都可能在DDI机制中发挥关键作用,随机抽样可能会遗漏这些重要因素并阻碍预测性能。
相比之下,SumGNN提供了一种可学习的方式来提取邻域的有用信息。此外,以往的工作都集中在二分类DDI预测上,而SumGNN则是在多类型关系网络上进行评估。
2.2 子图神经网络
GNN已被提出用于建模节点之间的关系,并已成功应用于各种领域。子图结构包含了许多图学习任务的丰富信息。例如,EGO-CNN应用局部自我意识(EGO)网络来识别图分类的结构。有人提出了一种多通道的子图分类方法。CLUSTER-GCN和GraphSAINT使用子图来提高GNN的可扩展性。与我们的工作更相关的是,有人将局部子图应用于链接预测,GraIL通过利用多关系信息将这一思想扩展到KGC任务。相比之下,SumGNN是由领域DDI预测问题驱动的,并首次对子图设计了图聚集模块,以获得可处理的路径。它还集成了新的多通道神经编码机制。
3.材料和方法
我们在这一部分介绍SumGNN(SumGNN代码可在https://github.com/yueyu1030/SumGNN)获得。我们在第3.1节聚集了问题设置,并在第3.2节详细描述了我们的方法。我们的方法可以分解为三个模块。
- 首先,我们提取药物对周围KG的局部子图,以获得有用的信息。
- 然后,我们提出了一个知识聚集方案来生成DDI的推理路径(即计算子图中任意一对相邻节点的边的信号强度分数,并且只保留一部分重要边)。
- 之后,我们描述了一个多通道知识数据集成模块(最终药物表示=知识聚集(由每一层的信号强度分数和相邻节点进行加权求和得到节点的关系感知消息,集成到同一层实体表示)+子图均值+化学指纹)来预测药效作用。
3.1 问题设置
3.1.1 定义1(药物相互作用图)
在给定药物\(\mathcal{D}\)和药效\(\mathcal{R}_D\)的情况下,药物相互作用图\(\mathcal{G}_{DDI}\)被定义为一组三元组\(\mathcal{G}_{DDI}=\{(u,r,v)|u \in \mathcal{D},r \in \mathcal{R}_D, v \in \mathcal{D} \}\),其中每个三元组\((u,r,v)\)表示药物\(u\)和药物\(v\)具有药效作用\(r\)。
3.1.2 定义2(外部生物医学KG表)
给定一组不同的生物医学实体\(\mathcal{E}\)和实体\(\mathcal{R}\)之间的生物医学关系,外部生物医学KG--\(\mathcal{G}_{KG}\)被定义为\(\mathcal{G}_{KG}=\{(h,r,t)|h \in\mathcal{E}, r \in \mathcal{R}, t \in\mathcal{E} \}\),其中每一项\((h,r,t)\)描述实体\(h\)和实体\(t\)之间的生物医学关系\(r\)。注意,我们将\(\mathcal{G}_{DDI}\)中的药物实体聚合到\(\mathcal{G}_{KG}\),即\(\mathcal{R}_D \in \mathcal{R}\)和\(\mathcal{D} \in \mathcal{E}\)。
3.1.3 问题1(多关系DDI预测)
DDI预测是在给定一对药物的情况下输出药效作用,从数学上讲,它是从一个药物对\((u,v) \in (\mathcal{D} \times \mathcal{D})\)中学习\(F \mathrm{:} \space \mathcal{D} \times \mathcal{D} \rightarrow \mathcal{R}_D\)到药效作用\(r \in \mathcal{R}_D\)的映射。
3.2 SumGNN方法
SumGNN由三个模块组成:子图提取、知识聚集和多通道知识数据集成。
- 对于给定的药物对,我们提取与KG中的药物对接近的潜在生物医学实体,形成子图。
- 然后,我们提出了一个新的GNN,它具有一个聚集方案,提供了一个用浓缩路径(即只保留一部分重要边)推理出DDI的机制。
- 给出这个路径图,我们进行多通道知识和数据集成,整合各种可用的信息源,以生成足够的药物对表示。
- 最后,采用解码分类器对交互结果进行预测。我们使用TransE初始化所有实体嵌入,其中实体被表示为\(\mathbf{h}_u^{(0)}\)(图1)。
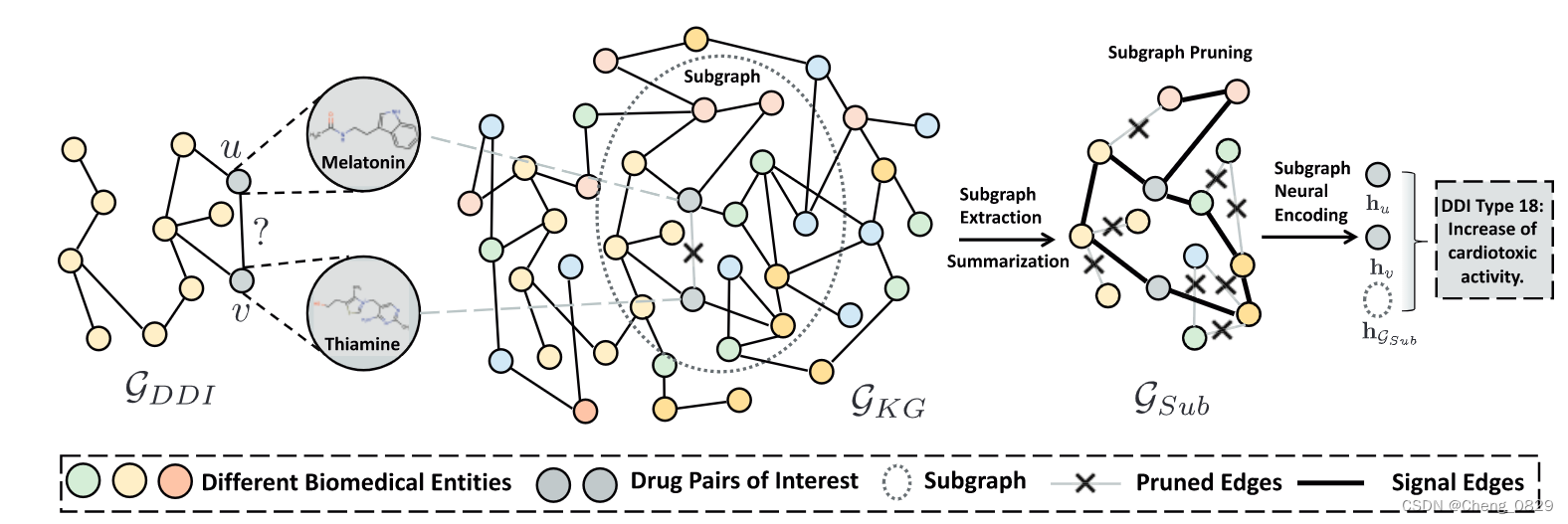
图1:模型结构
3.2.1 局部子图提取模块
生物医学KG描述了人类生物学的复杂机制。对KG中的几个药物对节点的调制可能会干扰相连的节点(例如,疾病、细胞成分等)。这会产生连锁反应,最终导致各种生理结果。随着药物对和生物医学实体之间距离的增加,这种影响会扩散。因此,为了理解DDI,我们关注药物对周围KG中的局部子图。
具体地说,对于药物对\(u\)和\(v\),我们首先提取\(u\)和\(v\)的\(k\)跳相邻节点\(\mathcal{N}_k(u)=\{s|d(s,u)≤k\}\)和\(\mathcal{N}_k(v)=\{s|d(s,v)≤k\}\),其中\(d(\cdot,\cdot)\)表示\(\mathcal{G}_{KG}\)上两个节点之间的距离。然后,基于这些结点的交集,我们得到了包围子图:\(\mathcal{G}_{Sub}=\{(u,r,v)|u,v \in \mathcal{N}_k(u) \cap \mathcal{N}_k(v),r \in \mathcal{R}\}\)。
根据前人的经验,节点相对位置对子图中中心节点\(u\)和\(v\)很重要,我们通过连接位置向量来增加嵌入到子图中的初始节点。对于子图\(\mathcal{G}_{Sub}\)中的每个节点\(i\),我们首先计算节点\(i\)与中心药物对节点\(u\)和\(v\)之间的最短路径长度\(d(i,u)\)和\(d(i,v)\),然后将其转换为位置向量\(\mathbf{p}_i=[\mathrm{one-hot}(d(i,u))\oplus \mathrm{one-hot}(d(i,v))]\)(即对于两个n位二进制编码的每一位取异或)。然后,我们将节点\(i\)的表示更新为\(\mathbf{h}_i^{(0)}=[\mathbf{h}_i^{(0)},\mathbf{p}_i]\)。
局部子图提取模块:即得到uv的包围子图\(\mathcal{G}_{Sub}\),并且找到各节点到子图中心节点uv的最短路径向量\(\mathbf{p}_i\),然后集成到各节点的表示\(h_i\)中。
3.2.2 知识聚集模块
知识聚集模块提供了一个用浓缩路径(即只保留一部分重要边)推理出DDI的机制。
除了提供预测结果之外,为了洞察生物学,我们还设计了一个知识聚集模块来将子图信息聚集成基于图的潜在DDI的路径。注意,这条路径不是一条直线,而是一个稀疏子图,因为产生DDI通常是由于许多类型的生物医学实体之间的复杂相互作用。我们需要保留信号最强的边,同时删除不重要的路径。为了实现这一点,我们采用了一种与层无关的、关系感知的自注意力模块来为\(\mathcal{G}_{Sub}\)中的每条边分配权重。这些权值是基于输入特征值\(\mathbf{h}^{(0)}\)生成的,并且表示用于剪除边的交互信号强度。
具体地说,我们将连接子图中任何一对相邻实体\(i\)和\(j\)的边的交互信号强度得分表示为\(\alpha_{i,j}\)。受transformer启发,我们使用自注意力机制,它考虑了子图中的所有邻居节点来生成注意力权重。这种注意力机制对我们来说是理想的,因为它在检查药物对周围的子图中的所有生物医学实体后生成信号强度分数。实体\(i\)和\(j\)的边的交互信号强度分数\(\alpha_{i,j}\)计算为:
信号强度分数\(\alpha_{i, j}^{(l)}\)衡量uv子图中任意两个实体节点之间的边的强度,是计算关系感知消息\(\mathbf{b}_v^{(l)}\)时的注意力权重,只通过\(ij\)的第一层表示进行计算。
知识聚集模块:设置阈值,剪除信号强度不够的边,即“浓缩路径”。
其中,\(\mathbf{W}^J\)和\(\mathbf{W}^I\)是包含子图中每个节点的表示的自注意力关键权重,\(r_{ij}\)对两个实体\(i\)和\(j\)之间的关系进行编码,\(\sqrt{d}_k\)是用于归一化的特征向量的大小,\(\gamma\)是信号阈值,并且\(\phi(x)=\frac{e^x-e^{-x}}{e^x+e^{-x}}\)是用于非线性变换的tanh函数。直观地说,该函数首先计算\(\mathbf{h}_i^{(0)}\)和\(\mathbf{W}^I\)之间的点积,从而得到节点\(i\)和子图中每个相邻节点之间的注意力分数,然后用关系嵌入法进行求和。按照同样的过程,通过\(\mathbf{h}_j^{(0)}\)与\(\mathbf{W}^J\)之间的点积,计算出节点\(j\)与相邻节点之间的注意力分数,然后用关系嵌入法进行求和。通过对\(i\)和\(j\)的每个相邻节点进行点积,最终得分考虑了所有子图信息。然后,经过非线性变换,计算出这条边的信号强度分数(范围从-1到1)。最后,我们应用一个阈值函数来筛选出强度得分阈值\(\gamma\)以下的边,设置它们的权重为0,因为它们对交互预测并不重要,设置为0会将这些边从GNN中的消息传递过程中剪除。此步骤应用于子图中的每条边。
现有的图形注意力方法为每一层中的每条边生成注意力权重。然而,这种方式可能会为同一条边提供跨层的潜在矛盾信号,从而阻碍可解释路径的生成。相比之下,SumGNN采用一种与层无关的注意力机制,只依赖于第一层嵌入来剪除边(只用\(h^{(0)}\))。它为模型的可解释性提供了一条明确的途径。由于许多生物网络是通过文本挖掘构建的,其中许多边可能是假负类,因此这种剪枝机制也能够降低噪声。
3.2.3 多通道集成模块
为了获得一个强大的DDI预测的表示,我们整合了一组不同的信息源。
3.2.3.1 通道1:知识聚集
使用我们上面描述的知识聚集方法,我们确定一个聚集子图,它对输入药物对很重要。我们希望利用子图为输入药物对生成潜在表示。我们使用以下消息传递方案对其进行集成。对于每个节点\(v\),我们类比注意力分数计算由第\(l\)层的信号强度分数\(\alpha_{u, v}^{(l)}\)进行加权得到关系感知消息\(\mathbf{b}_v^{(l)}\):
关系感知消息\(\mathbf{b}_v^{(l)}\)即节点\(v\)根据所有邻居节点\(u\)的表示\(h^{(l-1)}_u\)及uv之间的信号强度\(\alpha_{u, v}^{(l)}\)综合得到的消息。
其中\(\mathcal{N}_v\)表示子图\(\mathcal{G}_{Sub}\)中节点\(v\)的邻居,\(\mathbf{W}_r^{(l)}\)是用于转换第\(l\)层中节点\(u\)和\(v\)的隐藏表示\(\mathbf{h}_u^{(l)}\)和\(\mathbf{h}_v^{(l)}\)的权重矩阵。
为避免过拟合,我们使用基分解将\(\mathbf{W}_r^{(l)}\)分解为几个基矩阵\(\left\{\mathbf{V}_b\right\}_{b \in B}\)的线性组合:
其中,\(a\)是系数矩阵。然后,我们将关系感知消息\(\mathbf{b}_v^{(l)}\)集成到节点\(v\)的表示\(\mathbf{h}_v^{(l-1)}\),得到更新表示\(\mathbf{h}_v^{(l)}\):
节点\(v\)的表示\(h_v\)通过结合每层的关系感知消息\(\mathbf{b}_v^{(l)}\),从第1层到第\(l\)层逐步扩散。
其中\(\mathbf{W}_{\text {self }}\)是用于转换节点嵌入本身的权重矩阵。
3.2.3.2 通道2:子图特征
为了获得子图嵌入\(\mathbf{h}_{\mathcal{G}_{\text {sub }}}\),我们在第\(l\)层通过线性投影对子图\(\mathcal{G}_{Sub}\)中的所有节点\(i\)的嵌入取平均值:
3.2.3.3 通道3:药物指纹
化学指纹等分子信息已被证明是DDI的有力预测指标。因此,除了网络表示之外,我们还获得了摩根分子指纹\(\mathbf{f_v}\),这是每个药物\(v\)的预测描述符。值得注意的是,在KG中使用该特征作为输入节点特征是不可行的,因为KG包含了药物以外的各种类型的节点(例如副作用、疾病和基因),并且它们不能由摩根分子指纹表示,这导致GNN传播的节点特征不一致。
3.2.3.4 层次化的通道聚合
为了组装通过每一层生成的各种表示,我们采用了层聚合机制。我们连接每一层的节点嵌入和子图嵌入,即\(\mathbf{h}_v=[{\mathbf{h}^{(1)}_v},{\mathbf{h}^{(2)}_v},\cdots,{\mathbf{h}^{(L)}_v}]\)和\(\mathbf{h}_{\mathcal{G}_{Sub}}=[{\mathbf{h}^{(1)}_{\mathcal{G}_{Sub}}},{\mathbf{h}^{(2)}_{\mathcal{G}_{Sub}}},\cdots,{\mathbf{h}^{(L)}_{\mathcal{G}_{Sub}}}]\),其中\(L\)是层大小。为了整合化学指纹,我们通过串联化学表示\(\mathbf{h}_v=[\mathbf{h}_v \oplus \mathbf{f}_v]\)来更新层聚合嵌入。
最后,我们将不同的通道组合在一起,得到了输入的药物对表示\(\mathbf{h}_{u,v}=[\mathbf{h}_u,\mathbf{h}_v,\mathcal{G}_{Sub}]\)。为了预测关系,我们获得预测概率向量\(\mathbf{p}_{u,v}\),其中向量中的每个值对应于关系的合理性。通过将药物对的表示输入到由\(\mathbf{W}_{pred}\)参数化的解码器来计算\(\mathbf{p}_{u,v}\):
3.2.4 训练和推断
- 对于多分类(包括二分类)任务,我们对每条边\((u,r,v)\)采用交叉熵损失\(\ell_{CE}\):
其中\(\hat{y}_r=\operatorname{softmax}\left(\mathbf{p}_{u, v}^r\right)=\frac{\exp \left(\mathbf{p}_{u , v}^r\right)}{\sum_{i=1}^R \exp \left(\mathbf{p}_{u, v}^i\right)}\),\(y_r \in (0,1)\)是二进制指标,用来判断是否分类正确。
- 对于多标签分类任务,给定三元组\((u,r,v)\),我们采用二元交叉熵损失\(\ell_{\mathrm{BCE}}\)为:
其中\((u, r, w)\)是负采样得到的关系\(r\)的边。 这是通过将节点\(v\)替换为节点\(w\)来实现的,该节点根据分布\(P_w(v) \propto d_w(v)^{3 / 4}\)随机采样,\(\mathbb{E}\)表示符合相关分布的样本集的期望。
\(\hat{y}_r=\operatorname{sigmoid}\left(\mathbf{p}_{u, v}^r\right)\)和\(y_r^{u w}=\operatorname{sigmoid}\left(\mathbf {p}_{u, w}^r\right)\)分别是两条边的预测分数。 考虑所有边,SumGNN中的最终损失\(\ell\)为:
其中\(\ell\)采取等式 (7) 或 (8)中的哪种形式取决于任务类型。 在训练期间,我们通过使用随机梯度优化器(例如 Adam)最小化总损失\(\ell\)来学习模型参数.
在推理过程中,提取KG中未知的节点对(u,v)的子图,并按照相同步骤计算关系向量。对于多分类任务,我们选用概率最高的关系作为预测关系;对于多标签任务,我们收集所有关系的正负对应项的所有分数。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号