知识图谱-生物信息学-医学顶刊论文(Bioinformatics-2021)-MUFFIN:用于DDI预测的多尺度特征融合
2.(2021.3.15)Bioinformatics-MUFFIN:用于DDI预测的多尺度特征融合
论文标题: MUFFIN: multi-scale feature fusion for drug–drug interaction prediction
论文地址: https://www.researchgate.net/profile/Jianmin-Wang-3/publication/350100336_MUFFIN_Multi-Scale_Feature_Fusion_for_Drug-Drug_Interaction_Prediction/links/6063bb6592851cd8ce7ad5c1/MUFFIN-Multi-Scale-Feature-Fusion-for-Drug-Drug-Interaction-Prediction.pdf
论文期刊: Bioinformatics 2021
摘要
动机:
药物-药物不良相互作用(DDI)是药物研究的重要环节,是导致药物发病率和死亡率的主要原因。因此,识别潜在的ddi对医生、患者和社会都是至关重要的。现有的传统机器学习模型严重依赖手工特征,缺乏泛化能力。近年来,深度学习方法可以从分子图或药物相关网络中自动学习药物特征,提高了计算模型预测未知DDIs的能力。然而,以往的工作利用大量的标记数据,只考虑药物的结构或序列信息,而不考虑药物与其他生物医学对象(如基因、疾病和途径)之间的关系或拓扑信息,或考虑知识图(KG)而不考虑药物分子结构的信息。
结果:
为有效探索知识图中药物分子结构和语义信息的联合作用,提出了一种多尺度特征融合深度学习模型MUFFIN。MUFFIN可以结合药物自身结构信息和具有丰富生物医学信息的KG联合学习药物表征。在MUFFIN中,我们设计了一个包含交叉层和标量层组件的双层交叉策略,以很好地融合多模态特征。MUFFIN通过交叉从大规模KG和药物分子图中获得的特征,可以缓解深度学习模型中有限标记数据的限制。我们在三个数据集和三个不同的任务上评估了我们的方法,包括二分类、多分类和多标签DDI预测任务。结果显示,MUFFIN优于其他最先进的基准方法。
1.引言
药物-药物相互作用(DDI)引起的药物不良反应(ADR)可能会增加发病率和死亡率。因此,识别潜在的DDIS是至关重要的。近年来,人们采用了多种方法对DDI进行预测。预测DDIS的基本方法是传统的实验室方法。考虑到这些方法是劳动密集型的、耗时的和昂贵的,发现潜在的DDI的能力是非常有限的。因此,需要寻找准确可靠的计算方法。
机器学习是近年来兴起的一种计算方法,已被广泛应用于DDIS的预测。现有的基于机器学习的方法通过利用不同的与药物相关的相似性特征来预测潜在的DDiS,例如分子结构、副作用、显性相似性和基因组相似性。然而,这些作品在很大程度上依赖于手工特征和领域知识。最近的基于深度学习的方法可以从大量的数据中自动学习具有高度稳健性和泛化能力的抽象特征,缓解了传统机器学习带来的局限性。然而,以前的工作需要大量的标记数据,而这些数据可能会有假正类样本。它们通常要么关注药物的结构信息或SMILES序列而不考虑与药物相关的丰富语义信息,或者利用具有丰富生物医学信息的知识图(KG)而不考虑药物分子结构信息。
SMILES是指用ASCII字符串明确描述分子结构的规范,即把分子的三维模型,转换成二维图形,即常见的化学物质结构图。
虽然这些方法取得了较强的预测效果,但都没有考虑药物化学结构与KG之间的协同作用,从而限制了其预测能力。此外,大多数最新的工作考虑的是药物之间的相互作用的存在,将DDI预测作为一个二进制分类任务,而忽略了对药物之间特定类型的不良反应的重要研究。例如,KGNN确定了药物之间是否存在相互作用,而在我们的模型中,我们预测了这种相互作用的具体类型。例如,我们确定阿司匹林是否可以降低戈舍瑞林的排泄率,并可能提高血清水平。
考虑到上述局限性,我们提出了一种新的多尺度特征融合(MUFFIN)模型,这是一种利用药物化学结构和生物医学KG进行DDI预测的深度学习框架。我们设计了一种双层交叉策略,可以从基于卷积神经网络(CNN)的交叉层和标量层视角联合学习药物内部(化学结构)和外部(KG)特征的融合表示。该双层结构通过多粒度特征融合过程对多粒度特征进行有效组合,从而提高了DDI预测能力。此外,我们还在三种不同的DDI预测任务,即二分类任务、多分类任务和多标签任务上对MUFFIN模型进行了评估。实验结果表明,MUFFIN在三个任务上都取得了最好的性能,从而支持了KG的化学结构和知识特征相结合的意义。本文的主要贡献可以概括为:
- 我们提出了MUFFIN模型,这是一种新的基于深度学习的特征融合框架,用于二分类、多分类和多标签的DDI预测。它可以有效地将从药物分子结构中提取的特征与知识图相结合。
- 我们引入了包括交叉层和标量层模块的双层体系结构,可以用不同的粒度融合内部和外部特征。
- 交叉层模块通过对不同的特征进行向量积运算来提取和聚合局部特征(基于CNN)和全局特征;
- 标量层模块通过基于元素的乘积来提取许多细粒度的融合特征。
- 我们将MUFFIN与几个最先进的模型以及我们用于消融研究的模型的变体进行了比较。实验结果表明,我们的工作在三个不同的DDI预测任务上都超过了基准模型。
2.相关工作
近年来,人们提出了许多利用药物化学结构来解决DDI预测问题的工作。有人将DDI和药物结构相似性矩阵结合起来,生成DDI相互作用相似性矩阵,从而确定DDI候选者。有人利用药物表型、治疗、化学结构和基因组性质等四个相似性,并结合基于机器学习的五个模型(朴素贝叶斯、决策树、k近邻、Logistic回归和支持向量机)来处理DDI预测任务。有人利用药物化学结构的相似性作为特征,然后将药物-药物对输入深度神经网络(DNN)来预测相互作用类型。有人开发了一个端到端模型,该模型通过使用从药物SMILES序列中提取的子结构信息来预测DDIS来生成功能表示。
除了我们提到的基于药物化学结构的方法外,一些工作还利用了生物医学网络中药物的拓扑信息。有人提出了一个集合模型,该模型使用8种不同类型的药物数据和已知的DDI网络来预测DDI。有人发展了一个包含药物、靶点和副作用的图形卷积网络(GCN),并将DDI预测视为多关系链接预测任务。有人设计了一个具有注意力机制的多视图自动编码器,通过考虑已知药物属性的集成,如副作用、适应症和相互作用来预测DDIS。此外,对于KG中与药物相关的大型数据,有人将多源数据集集成到KG中,然后利用复杂的KG嵌入方法与卷积LSTM网络预测DDI。有人通过Bio2RDF工具将DrugBank和KEGG数据集转换为KG形式,然后利用选择性聚合邻居信息获得的表示来解决DDI预测问题。
然而,这些工作大多只是简单地考虑药物之间存在相互作用,而没有识别药物之间相互作用的副作用类型。同时,这些方法将结构信息和知识信息分开使用,没有考虑两者的互补作用。因此,我们的目标是设计一种新的融合策略,充分利用从药物分子图和大规模生物医学KG中提取的特征,生成适用于二分类、多分类和多标签DDI预测任务的药物表示。
3.模型方法
我们的问题表述在第3.1节中进行了总结;第3.2节介绍了我们提出的MUFFIN的框架;第3.3节描述了药物的两种表示;第3.4节开发了一种双层融合策略,该策略利用结构和与药物有关的知识信息来进一步学习药物表示;第3.5节表示如何利用这种表示法来准确预测DDI类型。
3.1 问题表述
在我们的研究中,我们将药物集合表示为\(D=\left\{d_1,d_2,\ldots,d_{N_d}\right\}\)及其对应的分子结构图集合为\(G_{drug}=\left\{g_1,g_2\right.\),\(\left.\ldots,g_{N_d}\right\}\),其中\(N_d\)是药物总数。对于二分类预测任务,我们定义了一个DDI关系矩阵\(Y\)用以表示药物\(d_i\)和\(d_j\)之间是否存在DDI,其中每个元素\(y_{ij}\in\{0,1\}\)表示存在\(d_i\)和\(d_j\)相互作用的实验证据(即\(y_{ij}=1\))或缺乏相互作用的证据(即\(\left.y_{ij}=0\right)\)。对于多分类预测任务,我们考虑DDI对的所有类型\(R_D\)(我们的工作中定义了81种类型的DDI关系)。对于多标签任务,考虑了200种不同的DDI类型。
我们将\(G_{kg}=\{(h,r,t)\mid h,t\in E,r\in R\}\)表示为\(\mathrm{KG}\),其中\(E\)表示实体集,\(R\)表示KG中的关系集。每个三元组\(\left(h_i,r_i,t_i\right)\)描述的是\(h_i\)和\(t_i\)之间存在以\(r_i\)为关系的连接(例如Loxoprofen、drug-target和COX2),其中\(h_i,t_i\in E,r_i\in R,i\in\left\{1,2,\ldots,N_{kg}\right\}\),\(N_{kg}\)表示KG中三元组的总数。
对于DDI预测问题,给定\(G_{kg}\)和DDI关系矩阵\(Y\)(或DDI交互对),我们旨在学习预测函数\(\hat{y}_{ij}=\mathcal{F}_1\left(\left(d_i,d_j\right)\mid\theta,G_{kg},Y\right)\)和一个把二分类和多分类分别从药物对\(\left(d_i,d_j\right)\)映射到特定类型的映射函数\(\mathcal{F}_2:D\times D\rightarrow\)\(R_D\),其中\(\theta\)表示模型参数,\(\hat{y}_{ij}\)表示药物对\(\left(d_i,d_j\right)\)之间相互作用的概率。
3.2 MUFFIN概述
MUFFIN的框架如图1所示。我们的框架由三个模块组成。
- 在表示学习模块中,我们采用消息传递神经网络(MPNN)和知识图表示方法(例如TransE)分别从分子图和知识图中提取分子结构特征和语义特征。
- 在特征融合模块中,我们设计了一个双层策略,包括交叉层和标量层单元。在交叉层单元中,我们将这两个特征交叉,然后使用CNN和flatten操作分别学习局部和全局特征。在标量层单元中,我们利用元素乘积来获得两个不同特征之间的细粒度交互特征。
- 在分类器模块中,我们将上述模块中学习到的特征串联起来,然后根据不同的分类任务使用各种分类器来预测DDI。接下来,我们介绍一下我们的框架的细节。
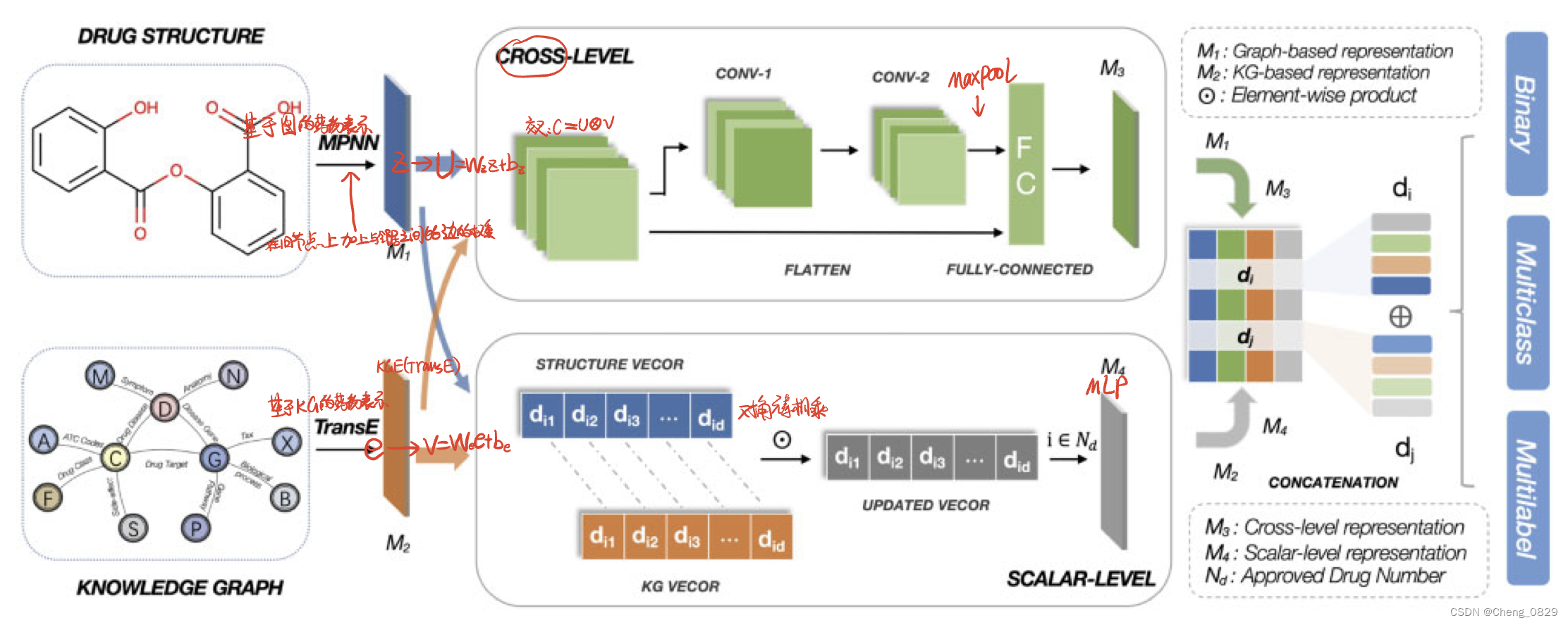
图1.MUFFIN框架:
1. 表示学习模块(左侧):MUFFIN利用药物结构信息(1维SMILES转换为2维药物分子图)并将其输入MPNN以学习基于图形的结构表示M1。对于KG中的实体,MUFFIN使用KGE方法(例如TRANSE)来获得基于KG的表示M2。
2. 特征融合模块(中间部分):上半部分实现交叉融合策略,两个表示作为叉积操作的输入,然后利用基于CNN的网络和flatten操作来获得局部和全局特征。通过级联操作和完全连接层来得到交叉表示M3。下半部分是标量层融合策略,它使用对角元素乘积来获得标量级表示M4。
3. 分类器模块(右侧):将四部分表示连接在一起,然后输入到全连接层,用于二分类、多分类和多标签DDI预测
3.3 表示学习模块
3.3.1 基于图形的表示
对于每种药物\(d_i \in D\),我们根据其SMILES序列构建了一个分子结构图\(g_i \in G_{drug},g_i=(\mathcal{V},\mathcal{E})\)其中\(\mathcal{V}\)代表原子,\(\mathcal{E}\)代表化学键。我们采用消息传递神经网络(MPNN)来生成\(d_i\)的结构表示。该过程涉及消息传递阶段和读出阶段。在消息传递阶段,我们执行了k次迭代,并通过聚合其邻居信息来更新节点\(p\)的表示。形式上,消息传递阶段生成节点\(p\)的表示可以描述如下:
其中\(U_{(k-1)}\)表示节点更新函数,\(M_{(k-1)}\)表示消息函数,\(N(p)\)表示图\(g_i\)中\(p\)的邻居,\(o_{pq}\in\)\(R^d\)表示节点\(p\)和节点\(q\)之间的边的表示,\(z_p^{(k-1)}\in R^d\)表示k-1次迭代后节点\(p\)的表示。消息函数定义为\(M\left(z_p,z_q,o_{pq}\right)=\mathcal{W}\left(o_{pq}\right)z_q\),其中\(\mathcal{W}(\cdot)\)是一个将边向量\(o_{pq}\)映射到\(d\times d\)矩阵的神经网络,即\(\mathcal{W}(\cdot)\)负责计算节点\(p\)和\(q\)之间的边的权值。基于图的最终药物表示\(z_i\in R^d\)可以在读出阶段通过对k次迭代后生成的节点表示求均值获得,读出阶段可以描述如下:
把药物\(d_i\)的分子结构作为一个图,求图中各节点的表示,然后迭代(即计算节点与其邻居之间的边的权重,然后加到新的节点表示中),最后求均值得到\(d_i\)的最终药物表示。
3.3.2 基于KG的表示
对于\(G_{\mathrm{kg}}\)中的每个实体(一种药物对应一个实体节点)和关系,我们使用一种广泛使用的KGE方法--TransE来获得基于KG的表示。具体来说,假设\(G_{kg}\)中存在三元组\((h,r,t)\),它通过优化平移规则\(\boldsymbol{e}_h+\boldsymbol{e}_r\approx \boldsymbol{e}_t\)来学习实体和关系嵌入,其中\(\boldsymbol{e}_h,\boldsymbol{e}_t,\boldsymbol{e}_r \in R^d\)。TransE模型经过训练来最小化基于边界的损失函数,如下所述:
让正样本距离\(f\left(\boldsymbol{e}_h, \boldsymbol{e}_r, \boldsymbol{e}_t\right)\)尽可能小,让负样本距离\(f \left(\boldsymbol{e}_{h^{\prime}}, \boldsymbol{e}_r, \boldsymbol{e}_{t^{\prime}}\right)\)尽可能大
其中\(\boldsymbol{e}_h,\boldsymbol{e}_r\)和\(\boldsymbol{e}_t\)分别是头部实体、关系和尾部实体的嵌入,\(f(\cdot)\)是TransE计算的距离,\(T\)和\(T'\)是三元组的正样本集合和负样本集合,\(\gamma\)表示边距参数。对于负类三元组\(T'\),它是通过用从\(G_{kg}\)中随机采样的实体和关系替换正三元组中的实体或关系来制定的。
3.4 特征融合模块
我们采用双层策略来融合基于结构图和KG的表示。融合特征用于表达多方面药物特征的交互信息。在双层融合操作之前,使用全连接层将特征向量转移到相同的公共向量空间\(\mathbf{u}\)和\(\mathbf{v}\)中。向量分别表示为由基于图的药物节点表示\(\mathbf{z}\)和基于KG的药物节点表示\(\mathbf{e}\)计算得到的\(\mathbf{u}\)和\(\mathbf{v}\)。具体来说,\(\mathbf{z}\)和\(\mathbf{e}\)代表所有药物的基于图形和KG的表示,如第3.3节所示。该过程可以表述如下:
其中\(W_z\)和\(W_e\)是可训练的权重,\(b_z\)和\(b_e\)是可训练的偏差。
3.4.1 交叉层
我们将转换后的基于图和KG的药物(\(d_i \in D\))表示分别设为向量\(u_i=\{u_{i1},u_{i2},\cdots,u_{id}\}\)和\(v_i=\{v_{i1},v_{i2},\cdots,v_{id}\}\)。\(u_i(v_i)\)表示向量\(\mathbf{u}(\mathbf{v})\)中的第\(i\)行。我们首先通过叉积运算构造了一个交叉矩阵\(C_i\),这个矩阵表达了\(u_i\)和\(v_i\)的交互作用如下:
其中\(C_i \in R^{d \times d}\),\(\otimes\)表示叉积操作符,\(d\)是药物向量的维度。然后我们使用带有池化层的CNN模型来学习局部交互特征,并压平交叉矩阵\(C_i\)来学习全局特征。我们将局部特征\(a_{il}\)和全局特征\(a_{ig}\)连接为[\(a_{il}||a_{ig}\)],然后将它们作为交叉特征\(a_i\)输入到全连接层中。具体过程可以描述如下:
其中\(W_c\)表示过滤器,\(*\)表示卷积算子,\(b_c\)表示偏置向量。在这里,我们分别采用ReLU和MaxPool作为非线性激活和池化函数。池化操作可以减少计算量和内存消耗,进而提高网络的表达能力。MLP是多层感知器。||表示连接局部和全局特征的连接运算符。
3.4.2 标量层
我们使用对角元素乘积运算对从基于图和KG的表示中学习到的\(u_i\)和\(v_i\)之间的特征交互进行编码,然后将对角元素向量输入到全连接层以获得标量级融合特征\(s_i\)。该过程描述如下:
其中\(s_i \in R^d\),\(\odot\)是对角元素乘积操作符。
3.5 分类模块
我们将四部分药物表示(包括基于图的表示\(u_i\)、基于KG的表示\(v_i\)、交叉表示 \(a_i\)和标量级表示\(s_i\))连接为药物\(d_i\)的最终表示。该过程可以描述如下:
其中\(x_i \in R^{4d}\)表示药物\(d_i\)的最终表示。
对于DDI预测任务,我们将一对药物\((d_i,d_j)\)的最终表示连接起来,然后将它们送入全连接层中,以预测DDI概率,如下所示:
其中\(\hat{y}_{ij}\)表示二分类预测任务中药物对相互作用的可能性,而在多类和多标签DDI预测任务中,它表示每种关系类型的概率分数。注意,在二分类和多标签任务中,\(\sigma\)是Sigmoid函数,而对于多类任务,\(\sigma\)是Softmax函数。
3.6 训练
在训练过程中,我们通过最小化二分类和多标签预测任务中的交叉熵损失来优化MUFFIN的参数,如下所述:
其中\(y_{ij} \in \{ 0,1 \}\)表示药物对\((d_i,d_j)\)在二分类任务和多标签任务中的互作用标签,\(y_{ij}\)的每个元素都是有200个元素(即200个DDI类型)的一个独热(one-hot)编码向量。对于多类别预测任务,损失定义如下:
独热(one-hot)编码:又称为一位有效编码,主要是采用N位二进制数来对N个状态进行编码,且任意编码不能是其他编码的前缀。例如“足球,篮球,排球,网球” -> “1000,0100,0010,0001”
其中\(N_c\)为多类DDI类型的个数,\(y_c \in \{ 0,1 \}\)描述当前类型\(c\)是否与样本对的真实标签相同,\(\hat{y}_c\)表示模型预测的样本\((d_i,d_j)\)属于类型\(c\)的概率。
多标签是指每条数据有多个标签。例如,学生所选的课程。因此多标签预测本质上就是多个相互独立且地位均等的二分类,因此也直接使用交叉熵损失函数。
多分类是指每条数据只有1个类别,和二分类一样,但是类别有多个,虽然也能视作多个二分类,但是彼此并不是相互独立且地位均等的二分类,因此不能直接使用交叉熵损失函数。

算法1: MUFFIN的训练算法。在算法1中,粗体字符表示所有药物的矩阵表示,例如\(\mathbf{C}=[\mathbf{C}_1,\cdots,\mathbf{C}_{i-1},\mathbf{C}_i,\mathbf{C}_{i+1} \cdots,\mathbf{C}_{N_d}]\),其中\(N_d\)为药物总数。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号