

科研绘图系列:R语言绘制微生物物种系统发育树(phylogenetic tree)
禁止商业或二改转载,仅供自学使用,侵权必究,如需截取部分内容请后台联系作者!
教程
教程内容
- 介绍
- 构成要素
- 有根树与无根树
- 构建方法
- 应用领域
- 说明的问题
- 加载R包
- 数据下载
- 导入数据
- 数据预处理
- 系统发育树可视化
- 准备画图数据
- 1.构建基础系统发育树
p1 - 2.添加条形图
p2 - 3.添加热图
p3 - 4.添加第二个热图
p4
- 保存PDF
- 总结
- 系统信息
介绍
物种系统发育树(Phylogenetic tree),也称为进化树或系统进化树,是一种以树状分支图形来表示各物种或基因之间的亲缘关系的图表。它利用生物的形态特征、分子序列(如DNA、RNA或蛋白质序列)等数据,通过数理统计算法来计算生物之间的进化关系,从而构建出一个反映物种进化历史的拓扑结构。
构成要素
系统发育树由节点(node)和进化分支(branch)组成:
- 节点:表示一个分类学单元,如属、种群、个体等。分支末端的节点对应一个基因或者生物体,代表实际观察到的最终分类。
- 进化分支:定义了分类单元(祖先与后代)之间的关系,一个分支只能连接两个相邻的节点。
- 分支长度:表示该分支在进化过程中的变化程度。标有分支长度的进化分支称为标度枝(scaled branch)。校正后的标度树(scaled tree)常常用年代表示,这样的树通常根据某一或部分基因的理论分析而得出。
有根树与无根树
系统发育树可以是有根的(rooted),也可以是无根的(unrooted):
- 有根树:有一个明确的根节点,表示所有物种的共同祖先。这种树可以清晰地显示物种的进化方向。
- 无根树:没有明确的根节点,仅表示物种之间的亲缘关系,不显示进化方向。
构建方法
构建系统发育树的方法主要有以下几种:
- 距离法(Distance-based methods):基于物种之间的进化距离,通过计算和比较序列之间的差异来构建树。常用的方法包括邻接法(Neighbor-Joining, NJ)和最小进化法(Minimum Evolution, ME)。
- 最大简约法(Maximum Parsimony, MP):寻找需要最少进化步骤的树,即假设进化过程中变化的次数最少。
- 最大似然法(Maximum Likelihood, ML):基于统计模型,寻找最有可能产生观测数据的树。
- 贝叶斯推断法(Bayesian Inference, BI):利用贝叶斯统计方法,通过计算后验概率来构建树。
应用领域
系统发育树在多个领域有广泛应用:
- 生物分类学:帮助确定物种的分类地位,构建生物的分类系统。
- 进化生物学:研究物种的起源、分化和进化历史,了解生物的进化过程。
- 生态学:研究物种的地理分布和生态适应性,了解物种在不同环境中的进化和适应机制。
- 分子生物学:研究基因家族的进化,了解基因的功能和进化历史。
- 医学:研究病原体的进化和传播,指导药物研发和疾病防控。
说明的问题
系统发育树主要说明以下问题:
- 亲缘关系:显示不同物种或基因之间的亲缘关系,帮助理解它们的共同祖先和进化路径。
- 进化历史:反映物种的进化历程,包括分化时间、进化速率和关键进化事件。
- 分类地位:确定物种在生物分类系统中的位置,为生物分类提供科学依据。
- 功能预测:通过比较基因或蛋白质的进化关系,预测其功能和结构特征。
- 生态适应:研究物种在不同生态环境中的适应性进化,了解生态系统的演变过程。
加载R包
安装ggtree的时候注意R包的依赖问题,耐心等待安装。
library(ape)
library(ggnewscale)
library(ggplot2)
library(ggtree)
library(ggtreeExtra)
library(RColorBrewer)
library(tidyverse)
# devtools::install_github("kevinwolz/hisafer")
library(hisafer)
数据下载
R语言系统发育树数据集下载链接:
- 百度网盘链接: 百度网盘下载链接
- 提取码:R语言绘制微生物物种系统发育树(phylogenetic tree)
导入数据
本次加载数据有四种数据类型,它们分别是
- 树结构文件:
pMAGs_bact_gtdtk_midroot.tree; - 物种信息:
pMAGS_tax.tsv; - 物种出现情况:
pMAGS_Presence_Datasets.txt; - 物种加和:
pMAGs_cov_sum.txt
BacTree <- read.tree("pMAGs_bact_gtdtk_midroot.tree")
dat <- read_tsv("pMAGS_tax.tsv")
dataset <- read_tsv("pMAGS_Presence_Datasets.txt")
covmax <- read_tsv("pMAGs_cov_sum.txt")
数据预处理
- 修改物种phylum水平的名称;
- 选择相对丰度前16的物种;
- 过滤物种类型
dat$p_c <- if_else(dat$p == "p__Proteobacteria", dat$c, dat$p)
dat$p_c <- gsub(".__", "", dat$p_c, perl = T)
dat$p_c <- gsub("_.$", "", dat$p_c, perl = T)
bacDat <- dat %>%
filter(d == "d__Bacteria") %>%
mutate(Abundance = 0.2)
mostAbundantTax <- bacDat %>%
group_by(p_c) %>%
summarise(total = n()) %>%
arrange(desc(total)) %>%
slice(1:16)
list <- mostAbundantTax$p_c
list <- c(list, "Nitrospirota")
bacDat <- bacDat %>%
mutate(p_c = if_else(p_c %in% list, p_c, "Other"))
bacDatset <- dataset %>%
filter(MAGs %in% bacDat$MAGs)
bacDatset$Busi <-
gsub("Busi", "Busi et al., Nat Com, 2022", bacDatset$Busi)
bacDatset$ENSEMBLE <-
gsub("ENSEMBLE", "Michoud et al, L&O, 2023", bacDatset$ENSEMBLE)
bacDatset$Tibet <-
gsub("Tibet", "Tibetan Glacier Genome and Gene", bacDatset$Tibet)
bacDatset$Tara <- gsub("Tara", "Tara Oceans", bacDatset$Tara)
bacDatset <- as.data.frame(bacDatset)
rownames(bacDatset) <- bacDatset$MAGs
bacDatset$MAGs <- NULL
bactcov <- covmax %>%
filter(MAGs %in% bacDat$MAGs) %>%
select(MAGs, count)
bactcov <- as.data.frame(bactcov)
rownames(bactcov) <- bactcov$MAGs
bactcov$MAGs <- NULL
系统发育树可视化
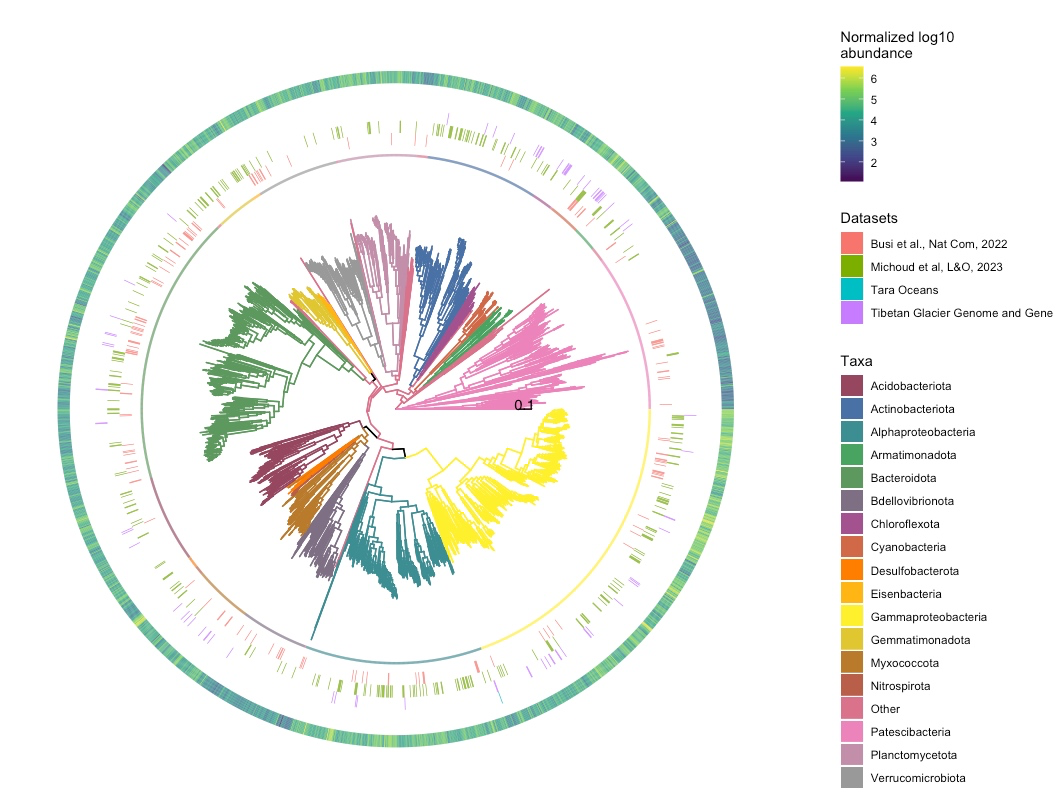
构建和美化了一个微生物物种的系统发育树,并添加了多个图层来展示不同的数据。
p1:基础的圆形系统发育树,节点颜色根据group变量着色。p2:在p1的基础上添加了条形图,展示每个节点的MAGs值。p3:在p2的基础上添加了热图,展示bacDatset数据。p4:在p3的基础上添加了第二个热图,展示bactcov数据。
准备画图数据
- 标准化物种的频次;
- 提取物种的树结构;
- 设置物种的颜色;
bactcov$count <- log10(bactcov$count)
a <- split(bacDat$MAGs, bacDat$p_c)
tree <- groupOTU(BacTree, a)
getPaletteBact <- colorRampPalette(brewer.pal(9, "Set1"))
bactColor <- getPaletteBact(length(unique(bacDat$p_c)) + 1)
bactColor[1] <- "black"
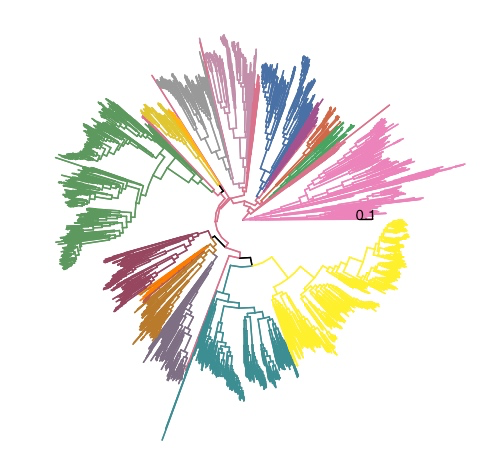
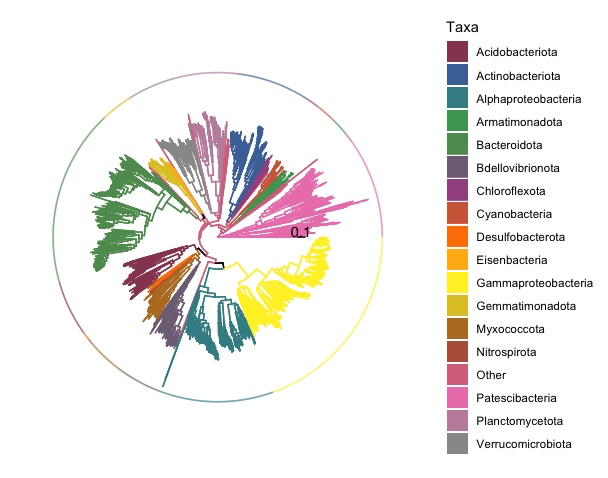
1. 构建基础系统发育树 p1
ggtree(tree, layout = "circular", aes(color = group)):使用ggtree函数创建一个圆形布局的系统发育树,节点颜色根据group变量进行着色。geom_tree():添加树的线条。theme_tree():应用ggtree的默认主题。geom_treescale(width = 0.1):添加一个比例尺,宽度为 0.1。scale_color_manual(values = bactColor, na.value = "transparent", guide = "none"):自定义节点颜色,缺失值透明,不显示图例。theme(legend.position = "right"):将图例位置设置在右侧。
p1 <-
ggtree(tree,
layout = "circular",
aes(color = group)
) + #
geom_tree() +
theme_tree() +
geom_treescale(width = 0.1) +
scale_color_manual(
values = bactColor,
na.value = "transparent",
guide = "none"
) +
# geom_text2(aes(subset=!isTip, label=node), hjust=-.3)+
theme(legend.position = "right")
p1
2. 添加条形图 p2
new_scale_colour()和new_scale_fill():重置颜色和填充尺度,以便添加新的图层。geom_fruit:在树的每个节点上添加条形图,数据来自bacDat,条形图的宽度为 0.01,高度为MAGs,填充颜色根据p_c变量。scale_fill_manual(values = bactColor[-1]):自定义条形图的填充颜色。labs(fill = "Taxa"):设置填充颜色的图例标签为 "Taxa"。
p2 <- p1 +
new_scale_colour() +
new_scale_fill() +
geom_fruit(
data = bacDat,
pwidth = 0.01,
geom = geom_bar,
mapping = aes(y = MAGs, fill = p_c, x = 1),
# orientation="y",
stat = "identity",
) +
scale_fill_manual(values = bactColor[-1]) +
labs(fill = "Taxa") +
new_scale_colour() +
new_scale_fill()
p2
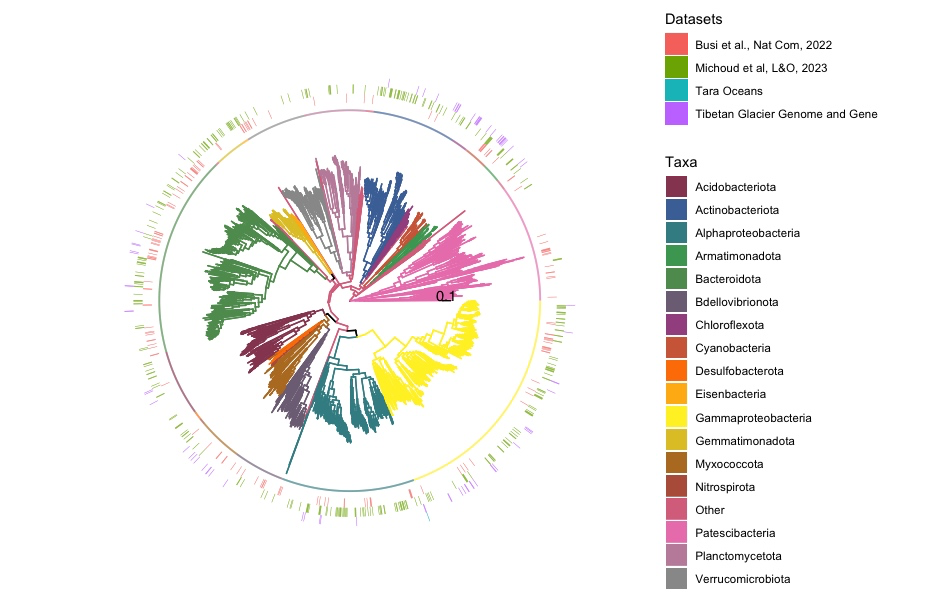
3. 添加热图 p3
gheatmap:在p2的基础上添加一个热图,数据来自bacDatset,热图宽度为 0.2,偏移量为 0.1,不显示列名,颜色默认。scale_fill_discrete(na.translate = FALSE):处理缺失值,不翻译为颜色。labs(fill = "Datasets"):设置填充颜色的图例标签为 "Datasets"。
p3 <-
gheatmap(
p2,
bacDatset,
width = 0.2,
offset = 0.1,
# offset=8, width=0.6,
colnames = FALSE,
color = NULL
) +
scale_fill_discrete(na.translate = F) +
labs(fill = "Datasets") +
new_scale_colour() +
new_scale_fill()
p3
4. 添加第二个热图 p4
gheatmap:在p3的基础上添加另一个热图,数据来自bactcov,热图宽度为 0.05,偏移量为 0.6,不显示列名,颜色默认。scale_fill_viridis_c():使用viridis色板填充热图。labs(fill = "Normalized log10\nabundance"):设置填充颜色的图例标签为 "Normalized log10 abundance"。
p4 <- gheatmap(
p3,
bactcov,
offset = 0.6,
width = 0.05,
colnames = FALSE,
color = NULL
) +
scale_fill_viridis_c() +
labs(fill = "Normalized log10\nabundance")
p4
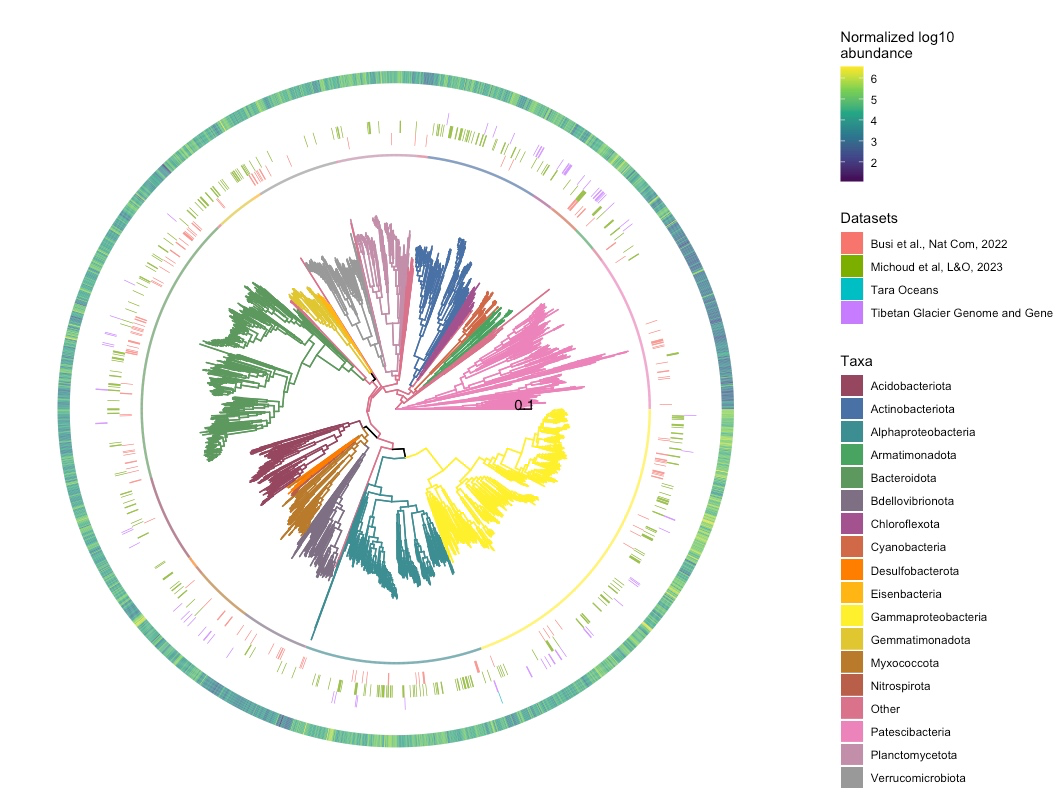
结果:微生物物种系统发育树图,它由以下部分组成:
- 最外层:物种的相对丰度值;
- 次外层:物种来自于的数据集分布;
- 次里层:物种的phylum水平;
- 最里层:不同分类物种的系统发育树结构。
保存PDF
将图片以PDF格式保存到本地
ggsave_fitmax <- function(
filename,
plot,
maxheight = 7,
maxwidth = maxheight,
units = "in", ...) {
if (is.null(plot)) {
return(FALSE)
}
dims <- get_dims(
ggobj = plot,
maxheight = maxheight,
maxwidth = maxwidth,
units = units
)
ggplot2::ggsave(
filename = filename,
plot = plot,
height = dims$height,
width = dims$width,
units = units, ...
)
}
get_dims <- function(
ggobj,
maxheight,
maxwidth = maxheight,
units = "in", ...) {
# Internal helper function:
# Treat all null units in a unit object as if they were inches.
# This is a bad idea in gneral, but I use it here as a workaround.
# Extracting unit names from non-atomic unit objects is a pain,
# so questions like "which rows of this table layout have null heights?"
# are hard to answer. To work around it, I exploit an (undocumented!)
# quirk: When calculating the size of a table layout inside a Grid plot,
# convertUnit(...) treats null units as zero.
# Therefore
# (convertHeight(grob_height, "in", valueOnly=TRUE)
# - convertHeight(null_as_if_inch(grob_height), "in", valueOnly=TRUE))
# does the inverse of convertUnit: It gives the sum of all *null* heights
# in the object, treating *fixed* units as zero.
#
# Warning: I repeat, this approach ONLY makes any sense if
# convertUnit(unit(1, "null"), "in", "x", valueOnly=T) == 0
# is true. Please check that it is before calling this code.
.null_as_if_inch <- function(u) {
stopifnot(packageVersion("grid") < "4.0")
if (!grid::is.unit(u)) {
return(u)
}
if (is.atomic(u)) {
if ("null" %in% attr(u, "unit")) {
d <- attr(u, "data")
u <- unit(
x = as.vector(u),
units = gsub("null", "in", attr(u, "unit")),
data = d
)
}
return(u)
}
if (inherits(u, "unit.arithmetic")) {
l <- .null_as_if_inch(u$arg1)
r <- .null_as_if_inch(u$arg2)
if (is.null(r)) {
args <- list(l)
} else {
args <- list(l, r)
}
return(do.call(u$fname, args))
}
if (inherits(u, "unit.list")) {
return(do.call(grid::unit.c, lapply(u, .null_as_if_inch)))
}
return(u)
}
if (inherits(ggobj, "ggplot") && !isTRUE(ggobj$respect) &&
is.null(ggobj$theme$aspect.ratio) && is.null(ggobj$coordinates$ratio) &&
is.null(ggplot2::theme_get()$aspect.ratio)) {
return(list(height = maxheight, width = maxwidth))
}
tmpf <- tempfile(pattern = "dispos-a-plot", fileext = ".png")
png(
filename = tmpf,
height = maxheight,
width = maxwidth,
units = units,
res = 120, ...
)
on.exit({
dev.off()
unlink(tmpf)
})
if (inherits(ggobj, "ggplot")) {
g <- ggplot2::ggplotGrob(ggobj)
} else if (inherits(ggobj, "gtable")) {
g <- ggobj
} else {
stop("Don't know how to get sizes for object of class ", deparse(class(ggobj)))
}
stopifnot(grid::convertUnit(grid::unit(1, "null"), "in", "x", valueOnly = TRUE) == 0)
known_ht <- sum(grid::convertHeight(g$heights, units, valueOnly = TRUE))
known_wd <- sum(grid::convertWidth(g$widths, units, valueOnly = TRUE))
free_ht <- maxheight - known_ht
free_wd <- maxwidth - known_wd
if (packageVersion("grid") >= "4.0.0") {
null_rowhts <- as.numeric(g$heights[grid::unitType(g$heights) == "null"])
null_colwds <- as.numeric(g$widths[grid::unitType(g$widths) == "null"])
panel_asps <- (
matrix(null_rowhts, ncol = 1)
%*% matrix(1 / null_colwds, nrow = 1))
} else {
all_null_rowhts <- (
grid::convertHeight(.null_as_if_inch(g$heights), "in", valueOnly = TRUE)
- grid::convertHeight(g$heights, "in", valueOnly = TRUE))
all_null_colwds <- (
grid::convertWidth(.null_as_if_inch(g$widths), "in", valueOnly = TRUE)
- grid::convertWidth(g$widths, "in", valueOnly = TRUE))
null_rowhts <- all_null_rowhts[all_null_rowhts > 0]
null_colwds <- all_null_colwds[all_null_colwds > 0]
panel_asps <- (matrix(null_rowhts, ncol = 1) %*% matrix(1 / null_colwds, nrow = 1))
}
panel_asps <- matrix(null_rowhts, ncol = 1) %*% matrix(1 / null_colwds, nrow = 1)
max_rowhts <- free_ht / sum(null_rowhts) * null_rowhts
max_colwds <- free_wd / sum(null_colwds) * null_colwds
rowhts_if_maxwd <- max_colwds[1] * panel_asps[, 1]
colwds_if_maxht <- max_rowhts[1] / panel_asps[1, ]
height <- min(maxheight, known_ht + sum(rowhts_if_maxwd))
width <- min(maxwidth, known_wd + sum(colwds_if_maxht))
return(list(height = height, width = width))
}
ggsave_fitmax("bacterialTree.pdf",
p4,
maxheight = 15
)
总结
本教程将指导你如何使用ggtree等一系列包在R语言环境中构建微生物物种的系统发育树。为了帮助读者更好地理解和应用,本教程提供了完整的数据和代码示例。
系统信息
R version 4.3.3 (2024-02-29)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.2
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Asia/Shanghai
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] hisafer_1.5.1 lubridate_1.9.3 forcats_1.0.0 stringr_1.5.1 dplyr_1.1.4
[6] purrr_1.0.2 readr_2.1.5 tidyr_1.3.1 tibble_3.2.1 tidyverse_2.0.0
[11] RColorBrewer_1.1-3 ggtreeExtra_1.12.0 ggtree_3.8.2 ggplot2_3.5.1 ggnewscale_0.4.10
[16] ape_5.7-1
loaded via a namespace (and not attached):
[1] yulab.utils_0.1.4 utf8_1.2.4 generics_0.1.3 ggplotify_0.1.2 stringi_1.8.3
[6] lattice_0.22-6 hms_1.1.3 digest_0.6.35 magrittr_2.0.3 timechange_0.3.0
[11] grid_4.3.3 fastmap_1.1.1 jsonlite_1.8.8 fansi_1.0.6 aplot_0.2.2
[16] scales_1.3.0 lazyeval_0.2.2 cli_3.6.3 rlang_1.1.4 munsell_0.5.0
[21] tidytree_0.4.6 withr_3.0.2 cachem_1.0.8 tools_4.3.3 parallel_4.3.3
[26] tzdb_0.4.0 memoise_2.0.1 colorspace_2.1-0 vctrs_0.6.5 R6_2.5.1
[31] gridGraphics_0.5-1 lifecycle_1.0.4 fs_1.6.3 ggfun_0.1.4 treeio_1.26.0
[36] pkgconfig_2.0.3 pillar_1.9.0 gtable_0.3.4 glue_1.8.0 Rcpp_1.0.12
[41] tidyselect_1.2.1 rstudioapi_0.16.0 farver_2.1.1 nlme_3.1-164 patchwork_1.3.0
[46] compiler_4.3.3


【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 阿里最新开源QwQ-32B,效果媲美deepseek-r1满血版,部署成本又又又降低了!
· 开源Multi-agent AI智能体框架aevatar.ai,欢迎大家贡献代码
· Manus重磅发布:全球首款通用AI代理技术深度解析与实战指南
· 被坑几百块钱后,我竟然真的恢复了删除的微信聊天记录!
· 没有Manus邀请码?试试免邀请码的MGX或者开源的OpenManus吧