paml正选择分析
自然选择是五种遗传力(突变、重组、选择、基因流、漂变)之一,选择压力分析更是进化分析中不可或缺的一项重要内容。
自然选择的检测
非同义替代与同义替换的比值,即:ω 值,也就是通常所说的dN/dS(或Ka/Ks)。
(1)当ω =1时,中性进化 (Neutral selection),即不受选择:
(2)当ω >1时,正选择(Positive selection);
(3)当 0 <ω <1时,负选择(Negative selection,也叫净化选择或纯化选择 Purifying selection)
不导致氨基酸改变的核苷酸变异我们称为同义突变,反之则称为非同义突变。
一般认为,同义突变不受自然选择,而非同义突变则受到自然选择作用。
在进化分析中,了解同义突变和非同义突变发生的速率是很有意义的。常用的参数有以下几种:同义突变频率 (Ks)、非同义突变频率(Ka)、非同义突变率与同义突变率的比值(Ka/Ks)。如果Ka/Ks>1,则认为有正选择效应。如果Ka /Ks=1,则认为存在中性选择。如果Ka/Ks<1,则认为有纯化选择作用。
dn/ds又叫ka/ks dN>dS 表示基因受到正选择:
杨子恒老师在他所著的《Computational Molecular Evolution》一书第8章第1节中这样写道:
With the synonymous rate used as a benchmark, one can infer whether fixation of nonsynonymous mutations is aided or hindered by natural selection. The nonsynonymous/synonymous rate ratio, ω = dN/dS, measures selective pressure at the protein level. If selection has no effect on fitness, nonsynonymous mutations will be fixed at the same rate as synonymous mutations, so that dN = dS and ω = 1. If nonsynonymous mutations are deleterious, purifying selection will reduce their fixation rate, so that dN < dS and ω < 1. If nonsynonymous mutations are favoured by Darwinian selection, they will be fixed at a higher rate than synonymous mutations, resulting in dN > dS and ω > 1. A significantly higher nonsynonymous rate than the synonymous rate is thus evidence for adaptive protein evolution.
翻译如下:
以同义突变率作为判定标准,可以推断非同义突变的保留是受到自然选择的支持还是阻碍。非同义突变率与同义突变率的比率,ω = dN/dS,可用以测量在蛋白质水平上的选择压力。
如果自然选择对基因的适合度没有影响,非同义突变将以和同义突变相同的速率被保留,即dN = dS,ω = 1。
如果非同义突变是有害的,净化选择将减小它们的保留速度,那么dN < dS,ω < 1。
如果非同义突变更受达尔文选择偏爱,它们将以大于同义突变的速度被保留,导致dN > dS,ω > 1。
非同义突变速度显著高于同义突变速度因此成为适应蛋白进化的证据。
CodeML中四个常见的模型假设及其工作流
在CodeML中,考虑不同序列间(考虑位点)或系统发育上的支系间(考虑支系Lineage)的ω 值不同,主要有以下四类常见的模型:
1. 枝模型(Branch model)主要用于对系统发育树中不同支系 ω值差异性进行界定,主要有三个模型:
(1)One-ratio model:假设系统发育树中所有支系的 ω 值相等;
(2)Free-ratio model:假设系统发育树中所有支系的 ω 值不相等;
(3)Two-ratio model:假设前景枝和背景枝的ω 值不同;
2. 位点模型 (Site model)主要假设数据集中不同氨基酸位点受的选择压力不同(而不考虑不同支系间受的选择压力差异)。
该模型主要用于检测正选择( ω >1)作用,共有8个不同假设的模型:
(1)M0(单一比率),即:One-ratio model,假设所有位点具有相同的 ω 值;
(2)M1a(近中性),假设仅有保守位点(0<ω <1)和中性位点( ω =1)而没有正选择位点( ω >1)存在,这两类位点的比率分别为p0和p1,其对应的ω 值分别为ω0、ω1;
(3)M2a(正选择),该模型在M1基础上增加了第三类ω值,即假设除了保守位点和中性位点外,还存在处于正选择压力下的位点( ω >1),这三类位点的比率分别为p0、p1和p2,其对应的ω 值分别为ω0、ω1和ω2;
(4)M3(离散),假设所有的位点ω 值呈简单的离散分布趋势;
(5)M7(beta),假设所有位点的 ω 属于矩阵(0, 1)并呈beta分布;
(6)M8(beta & ω ) ,该模型在M7基础上增加另一类ω 值(ω >1);
(7)M8a(beta & ω =1),与M8模型类似,但将ω 值固定为1(ω =0);
3. 枝位点模型 (Branch site model):主要假设不同氨基酸位点的和不同支系间受的选择压力均存在差异(既考虑位点间也考虑支系间的 ω 值存在差异),共有四个模型Model A、Model B、Model C和Model D,主要参数如下:
(1)Model A (Model 2, NSites=2, ncatG=ignored)
(2)Model B (Model 2, NSites=3, ncatG=ignored)
(3)Model C (Model 3, NSites=2, ncatG=ignored)
(4)Model D (Model 3, NSites=3, ncatG=2 or 3)
4. 进化枝模型 (Clade Model):与枝位点模型类型,能同时检测多个进化枝(Clade),共有CmC和 CmD 两种模型,主要参数如下:
(1)CmC (Model 3, NSites=2, ncatG=2 or 3)
(2)CmD (Model 3, NSites=3, ncatG=ignored)
CodeML 分析大致工作流简要如下:
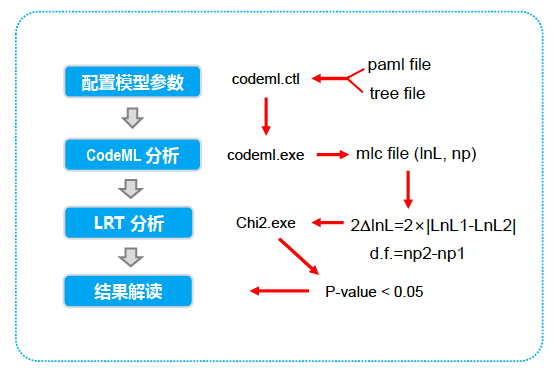
基因选择压力
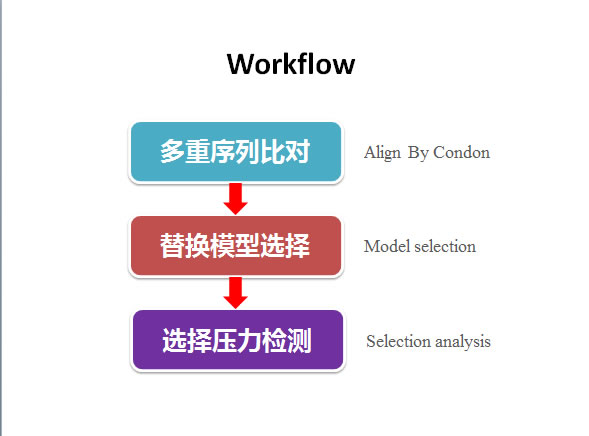
将序列基于密码子比对后,提交序列分析时,先选择一个最适的进化模型,再选择一个压力检测方法。
前者内置四种模型,包括Branch model、Branch-site model、Site model和Clade model,每个模型至少都有一组成对模型:零假设模型(Null model )和备选假设模型(Alternative model),其中Site model还内置三组成对模型(M0 vs. M3,M1a vs. M2a, M7 vs. M8)
杨子恒PAML选择压力分析之codeml(dN dS)
Codeml是PAML软件包下的一个程序,广泛应用于估算蛋白编码序列的同义替换和非同义替换速率,及检测序列是否受到正选择。
我们通过实例来分析某个编码序列氨基酸位点是否存在正选择作用。
安装好PAML, 准备序列比对文件,核酸序列得是3的倍数,序列中不能出现纯数字和特殊符号,但_可以,不能有额外的空格换行符等,
可用bioedit等工具将编码序列翻译后再进行比对,然后保存成phylip格式、Paml可识别phylip格式
树文件准备-任何可以正确描述序列文件中各序列文件的tree都可以,如果没有可以借鉴使用的树,可以用序列文件中的序列用Mrbayes, Phyml, PAUP等软件建树,先建立无根树
PhyML产生的树,格式可被Paml识别,Paup产生的树Nexus格式可能不能直接被Paml识别,可用figtree软件转存为Newick格式。
运行codeml程序,将序列比对文件和树文件同时拷贝到paml/bin文件夹下,或者确保codeml程序文件、codeml.ctl配置文件、比对好的XX.phy文件和序列文件、XX.tree树文件这四个文件处于同一文件夹中。
codeml.ctl配置文件的参数设置
seqfile 序列比对文件 treefile 树文件 outfile 输出文件 (任意命名,建议txt后缀便于打开)seqtype=1 clock=0 (使用无根树需设置为0) model=0
Nsites= 07 8 (0为单一参数模型,7 8 为相似模型,均允许局部替换率可以变化,两者有一个参数差异,其二者结果将用于LRST检验以保证结果的可靠性) 其余参数不做改变。
Model 0的结果里查找omega (dN/dS)的值,这里是0.29437 。(或者在分支模型计算结果里是几个omega的均值是最大且大于1的,且p值是显著的)
kappa是ts/tv
考虑单个氨基酸的dN/dS值,要先比较 Model7和model 8的Lnl (lnL )值,以确定哪一个模型更适合序列,然后再选择该模型的结果。
LRT = 2dl = abs(2 X (Lnl7-Lnl8)) (abs=绝对值)
第一步: 打开输出文件,找到Model7 和Model8两个Lnl 值相减,取绝对值,乘以2
第二步: 打开Paml附带的chi2程序,分别输入自由度和第一步的结果,自由度直接取1,因为M7,M8一个参数差异 输入:chi2 1 35,
即出现: df=1 prob = 0.02534=2.535e-02 得到的p小于0.05 (这里在branch model里最终结果序列的模型的p小于0.05 )
此处Paml用Naive Empirical Bayes (NEB) 分析检测显著性
CODONML in paml version 4.9f,
做选择压力分析的序列文件,需要先clustal和剔除终止密码子,这步可以在MEGA里完成。
1.MEGA7
MEGA7,是通过分别求取dn,ds的值,然后得到dn/ds。
将fasta格式的序列文件导入MAGA,
然后选择Distance——computer overall mean distance进入图一页面:仔细看下面的选项卡,在substitutions type 中选择 Syn-Nonsynonymous;Genetic code table 按照自己的序列选择;modle/method 选择 Nei-Gojobori method (No. of Differences);Substitutions to include 选择要计算的dn或者ds。下一步,就能得到dn或者ds,两者相比得到结果。
如果在distance——computer pairwise#####,然后按照后面步骤操作,结果会得到一个两两比较的矩阵(三角),我还不知道这个要怎么用。
如果只计算dn/ds,第一种应该够用了。
作者觉得视频教程里up主说做的是branch,然后取model=0是零假设,与我理解的有出入,我以为model=0是site,NSsite可以决定哪个是零假设哪个是备择假设。
posted on 2020-08-04 15:25 BPSO_mynotes 阅读(18583) 评论(0) 收藏 举报


 浙公网安备 33010602011771号
浙公网安备 33010602011771号