
RNA-Seq分析|RPKM, FPKM, TPM, 计算对比
在分析了若干转录组之后发现,处理数据的时候最重要的不是技巧多么绚丽,你调包的能力有多么强。而是把基本的概念特别是统计和数学上的方法咬烂嚼吐,才是真正理解和掌握了分析数据的底层原理:
在RNA-Seq的分析中,对基因或转录本的read counts数目进行normalization是一个extremely essential的过程,因为落在一个基因区域内的read counts数目取决于基因长度和测序深度。
Thats to say,一个基因越长,测序深度越高,落在其内部的read counts数目就会相对越多。
所以DE时,往往是在多个样本(样本来自不同组织、不同器官、不同个体、甚至做进化数据的时候是不同物种)中比较不同基因的表达量,如果不进行数据标准化,比较结果是没有意义的。
Therefore,我们需要标准化的two key factors 就是基因长度和测序深度,常常用RPKM (Reads Per Kilobase Million), FPKM (Fragments Per Kilobase Million) 和 TPM (Trans Per Million)作为标准化数值,前两者都是DESeq2 package中的funcitons。但是实践证明,在样本差异过大或者需要更加精准的比较或者定量目标基因的表达量的时候,还是TPM最为准确和有效。
来源于YouTube的一张示意图:

1、2、3样本total reads=35、45、106
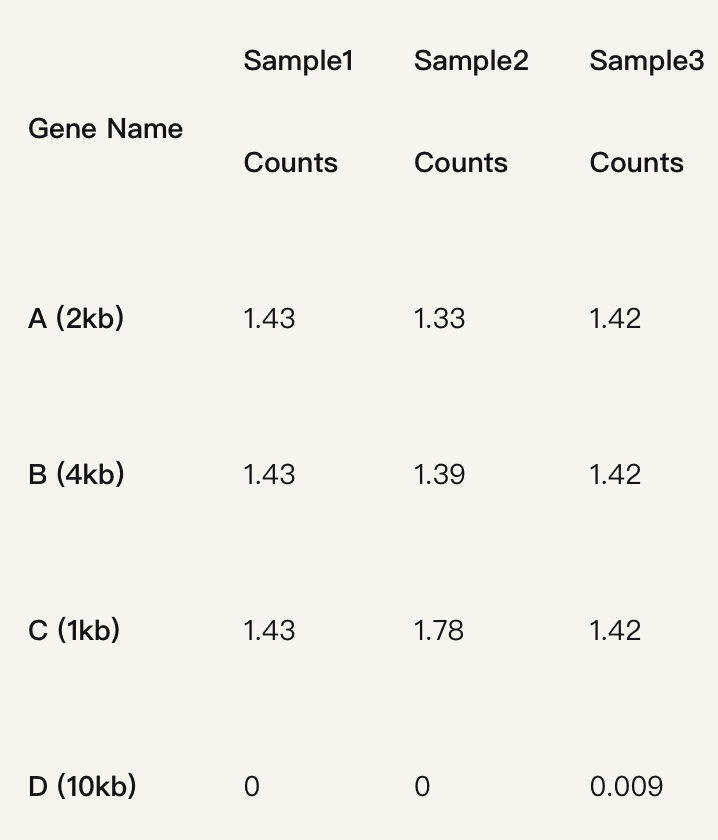
RPKM=10/35/2=1.43 (如下图所示)
而TPM的有效性在于它的处理基因测序的深度和长度的顺序是不同的。
即先考虑基因长度,再是测序深度:
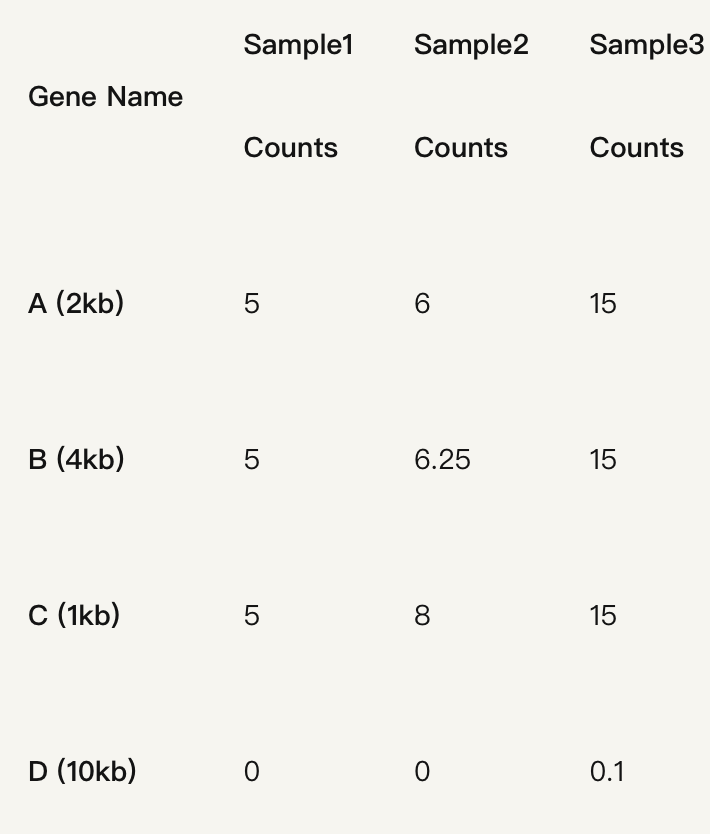

最后算出来的TPM=3.33
而再比对一下最后结果的total after normalized reads
RPKM:

TPM:

当我们看到这个结果的时候,就应该马上想到每个样本的TPM的总和是相同的,这就意味着TPM数值能体现出certain样本比对上target基因的reads的比例,而这个比例的总和在不同样本之间是相同的,所以可以使得该数值可以直接进行样本间的比较。

