
生信软件的安装和使用—— 建树前非保守序列的过滤
所有的事情,都是问题导向的,这里遇到的问题,便是怎么将多序列比对进行有效的过滤。
我们关注到了一个软件,bmge,这是一个不算很老的软件,怎么说呢,引用量还可以,文中对当前比较热的几个软件之间进行了对比 ,包括Gblocks,trimAI,和Noisy。
文章标题如下:
BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments
安装过程很简单:
conda install -c bioconda bmge
conda是怎么安装的不再赘述,前面的博客和公众号中讲过。
本软件调用的是java,如果之前conda已有Java,需要注意版本信息,防止影响其他工具的工作。
软件用法如下:
$ bmge -?
BMGE comes with ABSOLUTELY NO WARRANTY. This is free software, and you are welcome to
redistribute it under certain conditions. See the file COPYING.txt for details.BMGE (version 1.12) arguments :
-i <infile> : input file in fasta or phylip sequential format # 输入文件,建议用fasta格式,若为phylip格式,可以用 abyss phy2fa 做格式转换
-t [AA,DNA,CODON] : sequence coding in the input file (Amino Acid-, DNA-, RNA-, or CODON-coding sequences, respectively) # 输入文件格式,必填
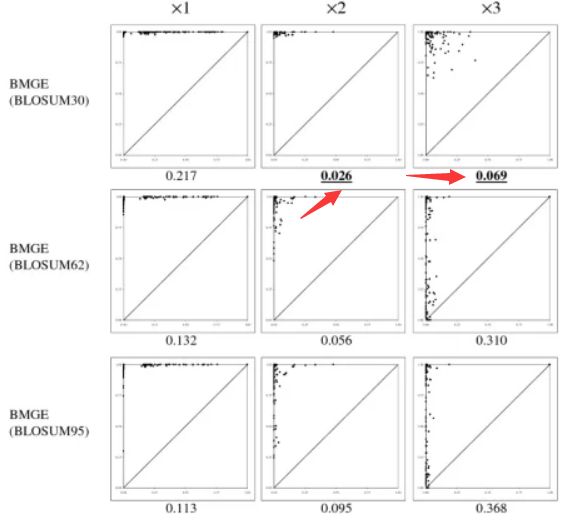
-m BLOSUM<n> : for Amino Acid or CODON sequence alignment; name of the BLOSUM matrix used to estimate the entropy-like value for each character (n = 30, 35, 40, ..., 60, 62, 65, ..., 90, 95; default: BLOSUM62) # BLOSUM矩阵,不支持DNA模式, 推荐使用30,详情看文献。
-m DNAPAM<n:r> : for DNA or RNA sequence alignment; name of the PAM matrix (n ranges from 1 to 10,000) and transition/transvertion ratio r value (r ranges from 0 to 10,000) used to estimate the entropy-like value for each character (default: DNAPAM100:2) # PAM 矩阵算法,其实我没看懂,就看到有人说选错了会造成误差放大,建议不用。。。用的话自己试试吧,默认的行最好。
-m DNAPAM<n> : same as previous option but with r = 1 # 同上
-m [ID,PAM0] : for all sequence coding; identity matrix used to estimate entropy-like values for each characters
-g <rate_max> : real number corresponding to the maximum gap rate allowed per character (ranges from 0 to 1; default: 0.2)
-g <col_rate:row_rate> : real numbers corresponding to the maximum gap rates allowed per sequence and character, respectively (range from 0 to 1; default: 0:0.2)-h <thr_max> : real number corresponding to the maximum entropy threshold (ranges from 0 to 1; default: 0.5)
-h <thr_min:thr_max> : real numbers corresponding to the minimum and maximum entropy threshold, respectively (range from 0 to 1; default: 0:0.5)
-b <min_size> : integer number corresponding to the minimum length of selected region(s) (ranges from 1 to alignment length; default: 5)
-w <size> : sliding window size (must be odd; ranges from 1 to alignment length; if set to 1, then entropy-like values are not smoothed; default: 3)
-s [NO,YES] : if set to YES, performs a stationarity-based trimming of the multiple sequence alignement (default: NO)
-o<x> <outfile> : output file in phylip sequential (-o, -op, -opp, -oppp), fasta (-of), nexus (-on, -onn, -onnn) or html (-oh) format; for phylip and nexus format, options -opp and -onn allow NCBI-formatted sequence names to be renamed onto their taxon name only; options -oppp and -onnn allow renaming onto 'taxon name'_____'accession number' (default: -oppp) # 输出文件
-c<x> <outfile> : same as previous option but for the complementary alignment, except for html output (i.e. -ch do not exist)
-o<y> <outfile> : converts the trimmed alignment in Amino Acid- (-oaa), DNA- (-odna), codon- (-oco) or RY- (-ory) coding sequences (can be combined with -o<x> options; default: no conversion) (see documentation for more details and more output options)上面密密麻麻的参数,去相关文献里面看,要么用了blosum参数,要么直接默认,真实效果还不知,因为我没有测试数据,tools的文献怎么说,当然说自己好啊,不信你看:
当然,文中毫不吝啬地说,其实,在分分歧度较低的时候,gblocks会好一些。
分歧度较低的时候,怎么样乱来都能建好树的好吧。
具体用着怎么样,我吃完了这个瓜会写在公众号里面的,希望这个瓜是甜的。
gblock的引用量很高,这里给出传送门,就不说怎么安装了:
Gblocks Documentation (csic.es)
Gblocks nad3.pir -t=p -e=-gb1 -b4=5 -d=y # 这是个例子,蛋白的。
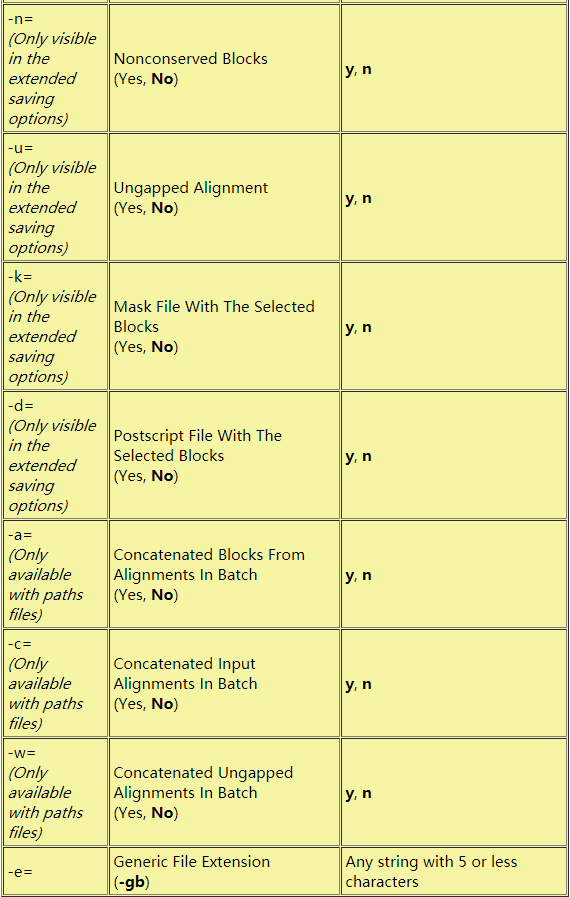
参数直接投喂在这里了:
请自行食用。
以上,
abysw
有个做比对的链接,有兴趣的同学可以试试Gblocks (vardb.org)



posted on 2020-11-30 19:43 Yuan-SW-F(abysw) 阅读(1507) 评论(0) 收藏 举报

 浙公网安备 33010602011771号
浙公网安备 33010602011771号