
生物信息-McScan(Python-jcvi)共线性画图
比较基因组学中,共线性的分析的图无疑是最漂亮的。
共线性分析可以很好地解释进化关系和多倍化事件。
本文主要介绍的是唐老师的Python版McScan(jcvi工具包),这个包很强大,但是其功能在官网的说明并不详细,在众人的博客中也比较零散。
我刚使用这个包的时候(2017年)还很难安装,需要预装各种依赖,不过现在的同学们很幸福了,可以直接用pip一键安装了。
软件包链接:https://github.com/tanghaibao/jcvi
安装过程很简单:
pip install jcvi
pip install git+git://github.com/tanghaibao/jcvi.git
如果安装不成功,再执行一次上述命令即可。
python 用conda安装即可。
官方配图如下:
![]()
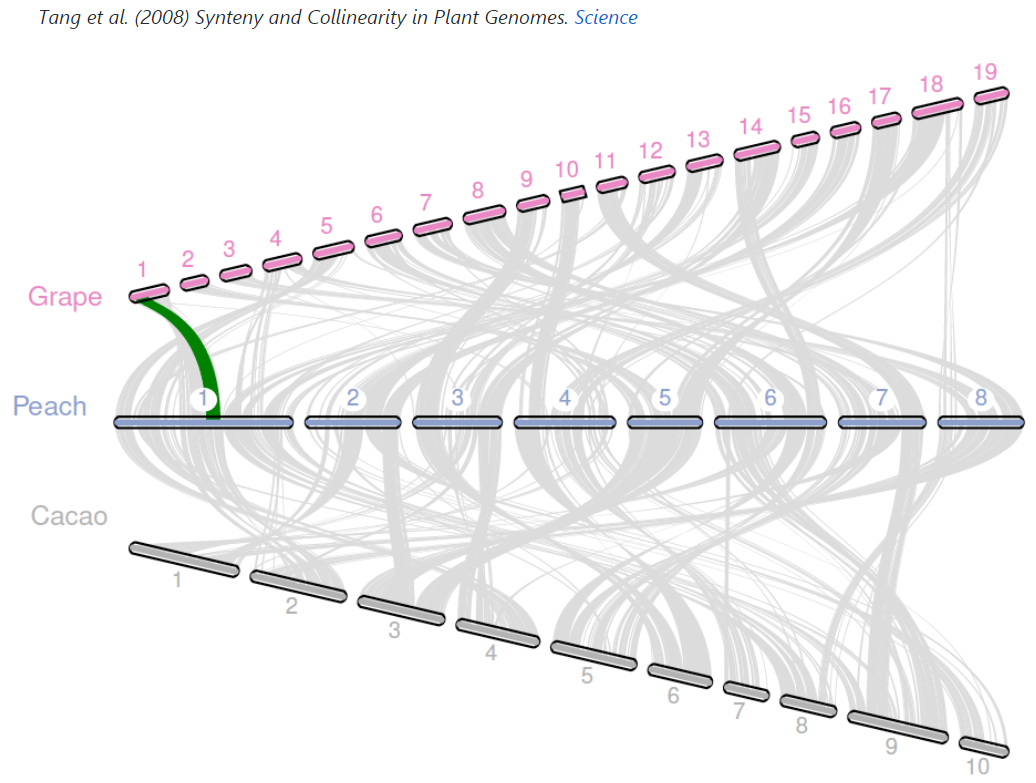
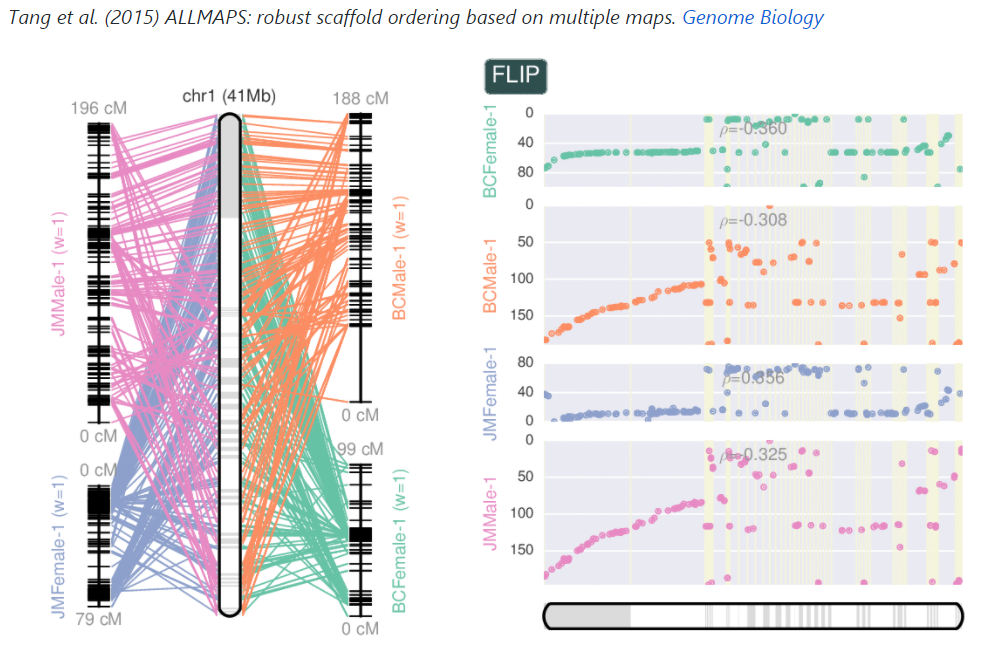
鄙人拙作如下:

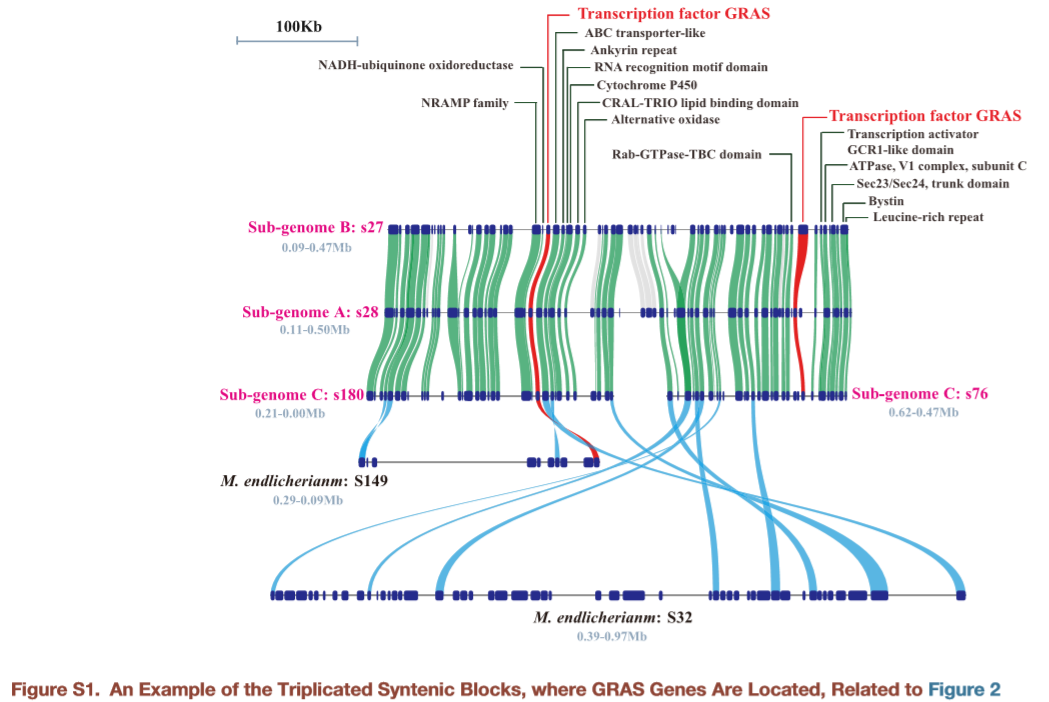
本文其实并没有想用常规的方法告诉同学们怎么用,我只是想告诉同学们一键生成最终结果的办法:
1. 两两物种之间的共线性分析和画图:
诸君只需要准备好下载好的两个需要比对的基因组序列文件和注释文件(species.gff),进行格式化:
#下载基因组相关数据
get-genome.pl
#格式化基因组序列文件
format_fa.pl
#格式化基因组注释文件
format_gff.pl
#共线性画图
#统计共线块的分布情况
2. 多物种基因组序列比对,保守序列/区域画图(准备好lastz软件,不需要准备注释文件)
# 获得物种两两比对结果
lastz-axt.sh reference species
# 获得多序列比对结果
roast tree *sing.maf roast.maf
# 获得各物种与reference的比对矩阵
由于发现了其他网站的搬运工,本处将未发表内容暂时隐去了,可能一年后会和发表文章一起更新。
# 生成配置文件
# 画多序列共线性图(如下,可以直观看到倒位,缺失等重要信息,图中注释信息位置均可以进行调整)
python -m jcvi.graphics anchors seqids layout
3. 多物种基因共线性图(准备好blastp软件,需要gff注释文件)
# blastp比对
# 获取各物种与reference的RBH比对矩阵
# 生成配置文件(anchors文件由RBH矩阵替换)
# 画图
python -m jcvi.graphics anchors seqids layout
本操作流程节约了各种配置编辑和试错的时间和精力,增加了无注释文件或者非编码区(全基因组序列,而非仅基因区)的共线性分析。
注意事项:
1. 虽然可以conda一键安装python,pip一键安装jcvi,但是如果有依赖在尝试两次安装jcvi后依然无法自动安装,请手动安装。
2. 注意所有文件在第一步的时候进行严格地格式化,请文件中不要出现特殊字符,尽量只有数据/字母/下划线(原始文件只有两个,基因组序列文件和注释的gff文件)。
3. 如果要添加颜色,请在矩阵中加上注释(红色:r*;黄色:y*)
*. 有任何BUG,请及时与管理员联系。
脚本将陆续公布于网站cospure.cn和github中。
脚本的写作和解读将陆续在微信公众号中进行讲解。
本博客主要用于文章和软件的前期撰写和测试,后期整理的详细版请关注微信公众号swxxfxxx。
以上,
abysw
![]()

请关注:
微信公众号:生物信息分析学习(swxxfxxx)
个人网站:http://cospure.cn/blog; http://undef.cn
博客园:https://www.cnblogs.com/abysw
posted on 2020-10-17 21:39 Yuan-SW-F(abysw) 阅读(11472) 评论(0) 收藏 举报

 浙公网安备 33010602011771号
浙公网安备 33010602011771号