Fastq/Bam的downsample
Fastq/Bam的downsample
本文作者:Sunny-King
发布时间:2022-08-08 16:10:54 星期一
本文链接:https://www.cnblogs.com/Sunny-King/p/Bioinformatics-Downsample.html
最艰难处理下机数据时,遇到了降采样的问题。目前有两个方向,一个是直接从fastq采样,另一个是从比对后的bam文件采样,节省了采样后再次比对的时间。下面介绍一下最近用到的一些工具。
一、Fastq
1、seqtk
Seqtk 是一种快速轻量级的工具,用于处理 FASTA 或 FASTQ 格式的序列。 它可以无缝解析 FASTA 和 FASTQ 文件,这些文件也可以通过 gzip 进行压缩。其中的seqtk sample可以用来从fastq中采样。具体用法如下:
# 对于pair-end数据需保证R1/R2的随机种子相同
seqtk sample -s 100 R1.fq.gz 0.5 > R1.0_5.fq.gz
seqtk sample -s 100 R2.fq.gz 0.5 > R2.0_5.fq.gz
可以指定一个整数提取一定数目的reads,也可以指定一个小数提取一定比例的reads。
二、Bam
1、samtools
samtools view提供了一个参数可以用于downsample,方法如下:
samtools view -s seed.freq input.bam -b -o downsample.bam
- 其中seed表示随机种子int类型的整数;freq为downsample的频率,float类型的小数。100.5表示以随机种子为100,取出50%的数据。
2、Picard DownsampleSam
该工具对SAM或BAM文件应用下采样算法。该算法中Pair-end的reads要么都保留,要么都丢弃。标记为non-primary的reads pair取将全部丢弃。每次读取被保留的概率为P,因此,使用完全相同的输入以相同的顺序和RANDOM_SEED的相同值执行的运行将产生相同的结果。
其使用方法与常规Picard的方法一致,Picard的一些公共参数该算法也包括。该算法主要有以下参数:
java -jar picard.jar DownsampleSam \
I=input.bam \
O=downsampled.bam \
P=0.5 \
R=100 \
ACCURACY=0.00001 \
STRATEGY=ConstantMemory
# --INPUT/-I, 指定输入bam文件
# --OUTPUT/-O, 指定输出bam文件
# --ACCURACY/-A, 算法的精度,误差尽可能保证在该精度范围,默认1e-4
# --RANDOM_SEED/-R, 设定随机种子,默认1
# --PROBABILITY/-P, downsample的比例,默认1.0
# --STRATEGY/-S, 采用策略,默认ConstantMemory
使用STRATEGY选项可以支持许多不同的下采样策略:
- ConstantMemory:使用哈希投影策略在固定内存中运行。适用大量的输入。准确性会随着输出数据的降低而降低。
- HighAccuracy:尽可能的保证准确性,即做到降采样的比例接近约定的比例。因此,该策略需要与输入的数据流中模板名称数量成比例的内存,因此在运行大型输入文件时将需要大量内存。适用于较小的数据输入
- Chained:是一个折衷的策略,综合了ConstantMemory和HighAccuracy的一些优点。使用ConstantMemory策略downsample到大约期望的比例,然后使用HighAccuracy策略精确完成。在一次传递中工作,将提供接近(但往往不如)HighAccuracy的精度,同时需要与从ConstantMemory策略到HighAccuracy策略发出的读取集成比例的内存。当对大的输入进行小比例的下采样时(例如,对数亿次读取进行下采样,只保留2%),效果很好。当输入包含>= 50,000个reads-pair时,应该有99.9%的准确率。适用于从大数据中取较低比例的数据。
3.两种方式的比较
我们测试了两种从bam中downsample的的方法,从相同的bam出发,设定不同的梯度和随机种子进行测试。
梯度设置为[0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9],随机种子设置为[1, 2, 3, 4, 5, 6, 7, 8, 9],每个组合设置两个重复。
首先计算了每个梯度的数据量与理论上的数据量,如下图所示。两种方法得到的数据与理论值具有很好的一致性。但是对于相同频率不同随机种子之间得到的数据量会存在细微的差距。
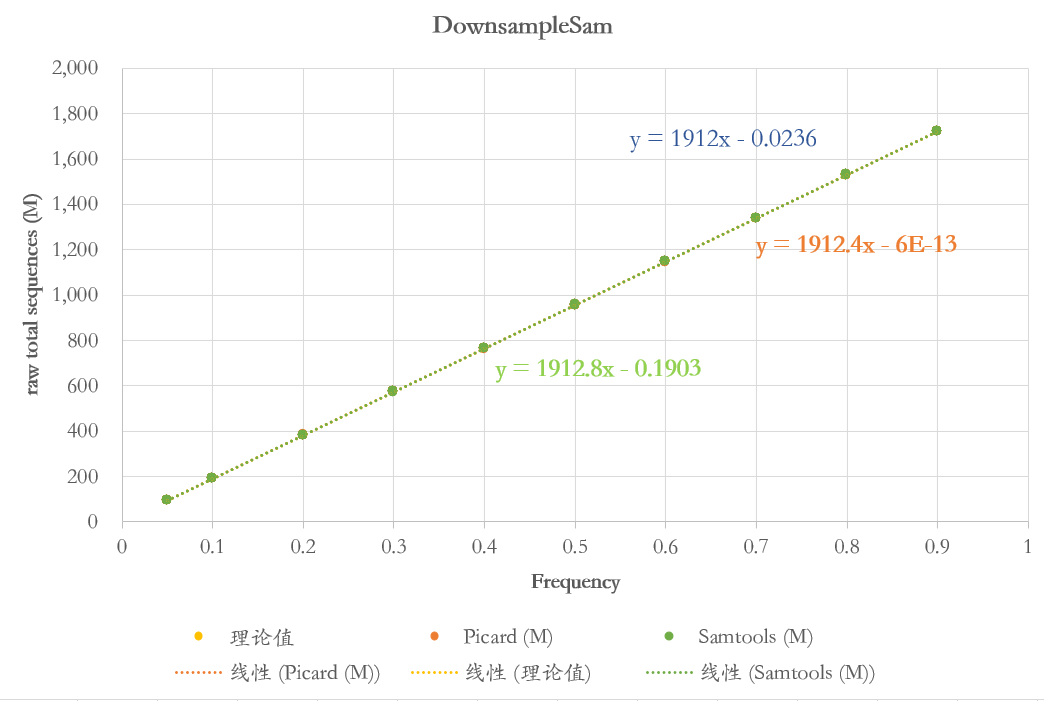
其次,比较了两个重复之间的一致性,发现设置相同的随机种子和频率时,得到的两个文件完全一致,MD5检验值相同。
最后,比较了时间上的差异,采用相同的计算资源,Picard要比samtools至少节约80%的时间。
本文作者:Sunny-King
本文链接:文章来源于博客园 https://www.cnblogs.com/Sunny-King/p/Bioinformatics-Downsample.html
转载要求:欢迎转载,转载之后请务必在文章明显位置标出原文链接和作者
错误修复:如有错误或疑问请联系博主
版权声明:本作品采用署名-非商业使用-禁止演绎 (by-nc-nd)许可协议进行许可
如果本文对您有帮助,请点个赞吧!志同道合的朋友可以点个关注。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号