
SHAPEIT算法简介
本文是基于SHAPIT2和SHAPEIT4的,先介绍SHAPEIT2的算法原理,然后简单介绍了一下SHAPEIT4更新的部分。文中介绍主要集中在算法部分,比较简介,具体内容请看参考文献。
[SHAPEIT2的参考文献为:a linear complexity phasing method for thousands of genomes;SHAPIT4的参考文献为:Accurate, scalable and integrative haplotype estimation]
相比于其他单体型分型软件,SHAPEIT2一共有两个改进,第一个改进是压缩,第二个改进是抽样。
压缩部分:
设定G为一个独立个体的基因型(genotype),H为除了这个个体以外的其他样本的单体型(haplotype),H和G涉及到的SNP相同。这里的H,说是其他个体的单体型,但是在计算刚开始的时候其他个体的单体型并不是已知的。所以在计算刚开始的时候是列举其他个体所有满足基因型G的单体型,然后从其中随机选取单体型作为已知的单体型,组成单体型集合H。在该情况下,推测独立个体的基因型,即推测出符合G的约束的两个单体型,即P(h|H)。

图1
在该方法中,对图H进行压缩,构建CHMM,即压缩隐马尔可夫模型。在原图H中,假设有K个单体型,涉及M个SNP,图2即压缩后的图Hg。压缩方法:在M个SNP中随机选择一个点作为起始点,给定一个压缩后的单体型个数J(J<M),在起始点左边,从左到右,逐个合并SNP,把在这些SNP范围内的单体型进行合并,知道合并后的单体型个数最接近J,这一部分算作一个片段(segment)。当合并后单体型个数最接近J时,断开segment,从接下来的一个SNP继续按此方法进行单体型合并,以此进行,直到把起始点合并进一个segment。在起始点的右边,从右向左进行合并,合并方法与左边类似。直到包含起始点。
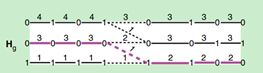
图2
在图Hg中,每个点和每个边均被赋予权重,权重分别为穿过该点和边的单体型的个数,分别记作c(km)和c(km,km+1)。每个点和边都至少有一个单体型穿过,并且H中的每个单体型在Hg中都有唯一路径。
压缩后的图去除了单体型的局部冗余,并且即使样本量增加或SNP增加,图的大小仍在一定范围内,图中的节点个数为JM个。完成压缩后,不再基于H图建立隐马模型,而是基于Hg建立模型,即P(h|hg)。则有M个隐藏状态z={z1,...,zm,...zM},和M个观测状态{h1,..,hm,...,hM},这M个观测状态即为Hg中表示的单体型。
基于Hg的隐马模型,其参数为:
起始状态概率:

状态转移概率:


状态发射概率:
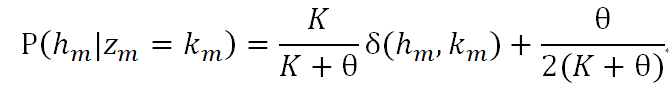
抽样部分:
对于一个无关个体,设G和D分别观测的基因型和未知的二倍体。目标就是求出P(D|G,hg)。
对于给定的G,如果G中含有s个杂合子位点,则所有单体型的个数为2s,当SNP个数增大时,该数量不可计算。所以使用类似压缩图的方法对其进行压缩,形成sg。通常sg 每个片段包含3个杂合位点。每个单体在sg中都有唯一路径x={x1,..,xm,...,xM}。
与图Hg相比,有sg两个不同,第一是片段的边界不同,第二是图sg没有权重。
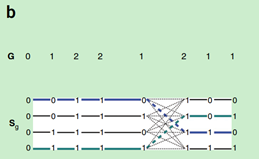
图3
P(D|G,hg)即从P(h|hg)中抽取中的一个路径h,也就相当于从P(x|hg)中抽取一个路径x放入到sg。即获得P(x|hg),其中x是sg中的一个路径。
根据马尔科夫链特性,即当前节点仅与前一个节点有关,则在给定节点xm时,xm-1和xm+1无关。所以有(这里有一个奇怪的特性,因为x是观测链,正常情况下x是不具有马尔科夫性的,但是这里却使用了马尔可夫性。这里可能是因为状态空间和观测空间是一致的,都是从{0,1}取值,所以也具有马尔可夫性,而且从文献可以看到在推测出观测链x之后,还有把x放入H进行下一个个体单体型的推测,这也要求其具有马尔可夫性):

这就构成了马尔可夫链,其中状态和转移分别为sg中的点和边。从P(x1|Hg)中随机抽取一个节点,然后使用P(xm|xm-1,Hg)进行迭代,直到完成M个节点的完整路径。
要计算P(x|Hg)即需要知道P(x1|Hg)和P(xm|xm-1,Hg)。另外我们可以看到P(x|Hg)其实就是P(x,z|Hg)的一个边缘概率。P(x,z|Hg)是联合概率,它有两个边缘概率,分别是P(x|Hg)和P(z|Hg)。联合概率和边缘概率的关系为:
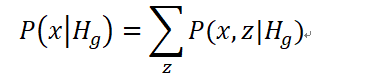
所以从解决边缘概率P(x|Hg)的问题转换为求联合概率P(x,z|Hg)。
对联合概率,有公式:
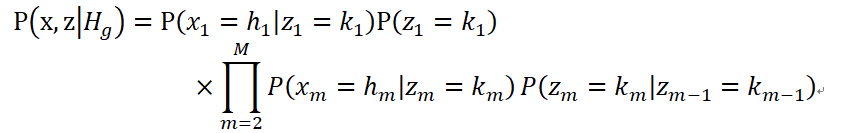
对于联合概率的求解,不是直接计算,而是采用前向后向的算法。
对于给定位置m,在m之前,使用前向算法:

在给定位置m之后,使用后向算法:
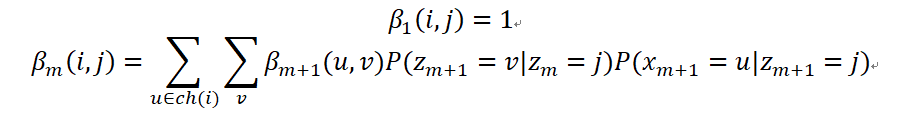
前向后向算法计算的是联合概率,所以最后使用联合概率的值计算边缘概率:

这里只获得了P(x1|Hg)和P(xm,xm+1|Hg),并没有获得P(xm+1|xm,Hg),这里是一个吉布斯采样的过程,基于吉布斯采样有一个特性,即联合概率正比于条件概率:

此时就获得了条件概率P(xm+1|xm,Hg)的近似值,当获得这两项之后就可以使用一下公式,抽取符合基因型G的单体型了。

这样就可以计算每个x的概率。对于所有符合G的单体型对,选出概率最大的既可。每计算完一个个体,就把算出的结果放入H,重新构成一个新的隐马模型,然后对下一个个体进行计算,如此迭代,直到计算完成。
SHAPEIT4更新内容:
SHAPEIT4更新了两个内容:1.更新H集合的构建方式;2.允许三种额外信息辅助分型。

图4
1.更新H集合的构建方式
在SHAPEIT2中是从所有样本可能的单体型中随机选取K个构成集合H,然后对H进行压缩,基于压缩后的图构建模型。在SHAPEIT4中,在除了当前样本以外的所有样本可能的单体型上,使用PBWT算法,选出与当前样本可能单体型最相似的P个单体型构成集合H,然后按照每8个SNP一组,对H进行SHAPEIT2类似的压缩,然后建立模型。
2.允许三种额外信息辅助分型
在SHAPEIT4可以使用三种额外信息来辅助进行SNP分析,这三种额外信息分别为:reference panel、haplotype scaffold和reads。使用这三种额外信息可以使分型结果更加准确。
其中S和R分别表示haplotype scaffold和reads提供的局部分型结果,reference panel的信息是在构建H时使用的,若存在reference panel,会在基于reference panel和其他样本的共同基础上建立PBWT算法,选出最相似的单体型,进而构建H。图5中S,即haplotype scaffold信息,在对图中Genotype graph构建的时候作为约束条件使用的。通过删除与haplotype scaffold不一致的候选h,可以减少Genotype graph的尺度。图5中的R,即reads信息,是在sampling的时候使用的,在进行sampling时,会允许选择的单体型h和reads信息有一定的错误率,并且通过选择的单体型h和reads信息进行比较,与reads信息越一致的单体型其权重越大,结果也更倾向于这样的单体型。根据以上描述,分别对reference panel、haplotype scaffold和reads信息进行利用,以辅助单体型分型。
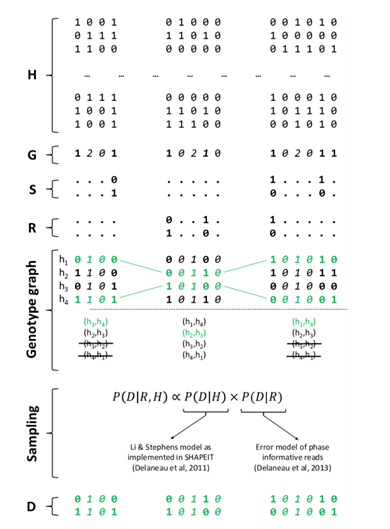
图5

