
svtools lmerge具体算法
svtools具有不同的子命令以实现不同的功能,其中一个就是lmerge。根据其帮助文档(merge LUMPY calls inside a single file from svtools lsort)可以看出,它是在lsort之后对一个vcf文件内的变异进行合并的,但只是知道它的功能而不知道它的原理,还是不能放心的使用它。所以就从它的代码看一下它是怎么操作的。
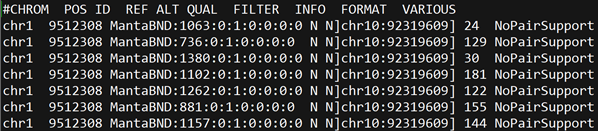
图1. lsort后的vcf文件,没行代表一个变异。
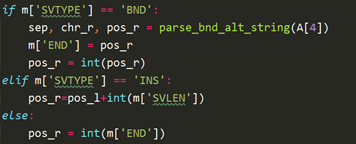
图2. 代码
由代码可知,它是对每个变异获得两个端点,一个就是vcf文件中的POS,把它作为第一个端点。然后获取第二个端点,根据变异类型不同,第二端点的获取方式也不同。如果变异类型是BND,则会解析vcf文件中REF位置的内容,获得其中的位置信息,作为第二端点。如果变异类型是INS的话,就把第一个端点加上SVLEN作为第二端点。其他情况获取vcf文件中END的信息作为第二端点。

图3. vcf文件展示
由于vcf文件每行内容都太长,所以上图换行展示。格外注意这里有CIPOS和CIEND两个信息。
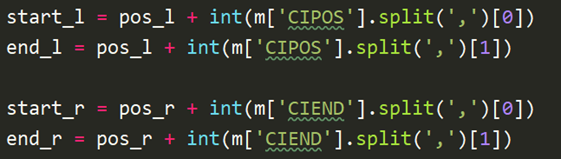
图4. 代码
由以上代码可以看出,在获取到两个端点之后,又分别根据CIPOS和CIEND的信息,把两个端点分别扩展成了两个区间。

图5. 把两个端点扩展为两个区间后的变异信息
可以看出,每个变异都含有两个区间,剩下的就是对这两个区间进行判断,两个变异记录是否为同一个变异。判断的标准:若这两个区间同时有重叠,则认为这两个变异记录是同一个变异(当然他们所对应的染色体也应该是一样的)。然后就可以对他们进行合并。

图6. vcf文件展示
vcf文件里有一个信息,PRPOS,这个信息是一系列数字,个数等于CIPOS[1] - CIPOS[0] + 1,表示第一端点处在每个位置的概率。另一个信息是PREND,也是一列数字,个数等位CIEND[1] - CIEND[0] + 1,表示第二端点处在各个位置的概率。
这里进行同一变异的合并,合并时,分别把第一区间和第二区间按照位置进行对齐,分别获得可以包含所有第一区间和第二区间的最大区间。例如一共有两个变异记录可能是同一变异,他们的第一区间分别为[start11=290 end11=390]和[start21=350 end21=580],则对齐后的区间为[start1=290 end2=580];若第二区间分别为[start12=680 end12=790]和[start22=780 end22=1000],则对齐后的区间为[start2=680 end2=1000]。按照区间的位置,也把PRPOS和PREDN进行对齐,前后空着的位置补0,然后把对齐后的PRPOS按位置进行相加,也把对齐后的PREND进行相加,就获得了与对齐区间长度一样的,PRPOS和PREND。然后找出PRPOS中数值最大的位置,然后对应到对齐后的第一区间,那就是变异合并后第一端点的位置。找到PREND中数值最大的位置,然后对应到对齐后的第二区间,那就是变异合并后第二端点的位置,这样就找到了变异合并后的两个端点的位置。
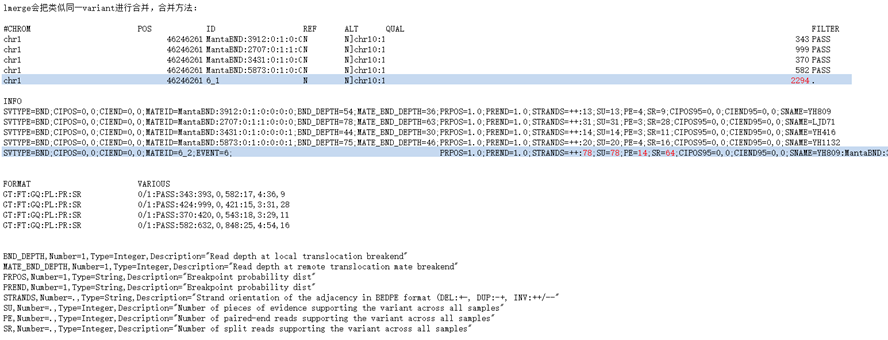
同时vcf文件里还会有QUAL、SU、PE、SR这些值,这些值的含义在vcf的header里都可以找到,他们的值合并的时候是直接相加的。这样就把代表同一变异的不同记录进行了合并,这就是svtools lmerge进行变异合并的原理。

