

GSEA
不太懂为什么现在生信分析这一部分都不喜欢写具体的原理,直接上来就是一堆怎么下载文件,怎么调用函数的操作,这篇文章想用通俗易懂的语言写一下原理部分,甚至贴一些代码。
今天看到的一个答案回答方式很喜欢
最简单的答案
主要是把基因列表降序排列,再来看待富集基因list上的基因,每出现一个,曲线就上升,没出现曲线就下降,计算曲线的最高点的值,背景分布就是把基因顺序打乱,然后同样计算一个类似的值。
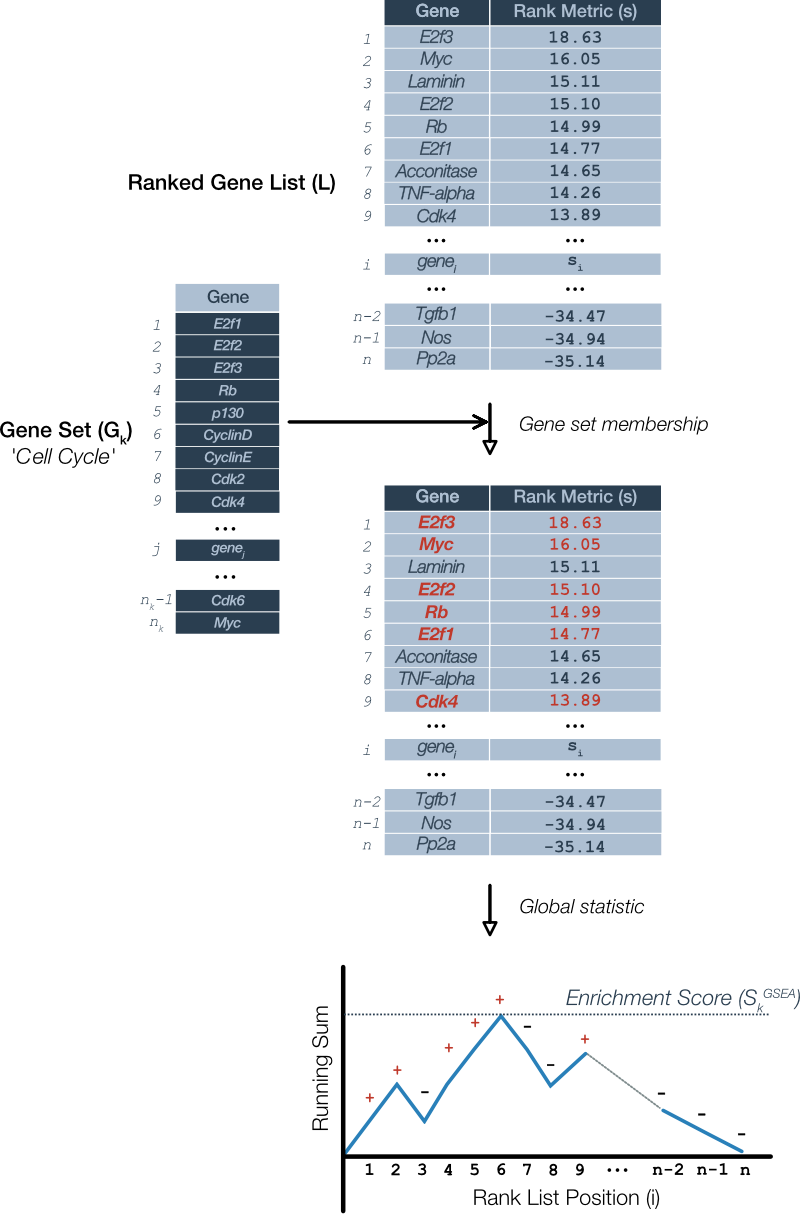
中等难度的答案

这部分就是直接看原理部分,非常漂亮的公式,大致就分为两部分,在待富集基因list上的比例,和不在待富集基因list上的比例,这两部分最后都是1,所以最后能够回到0的位置,也就是曲线其实是两个比例的差值最后是0.
至于这个alpha怎么选就得看更难得公式推导了,一共是分了两种选择,一种是0,一种是1,具体选择哪一种可以看参考文献2,写得非常得详细。
关于为什么得分会存在负数
说明富集在尾部呀
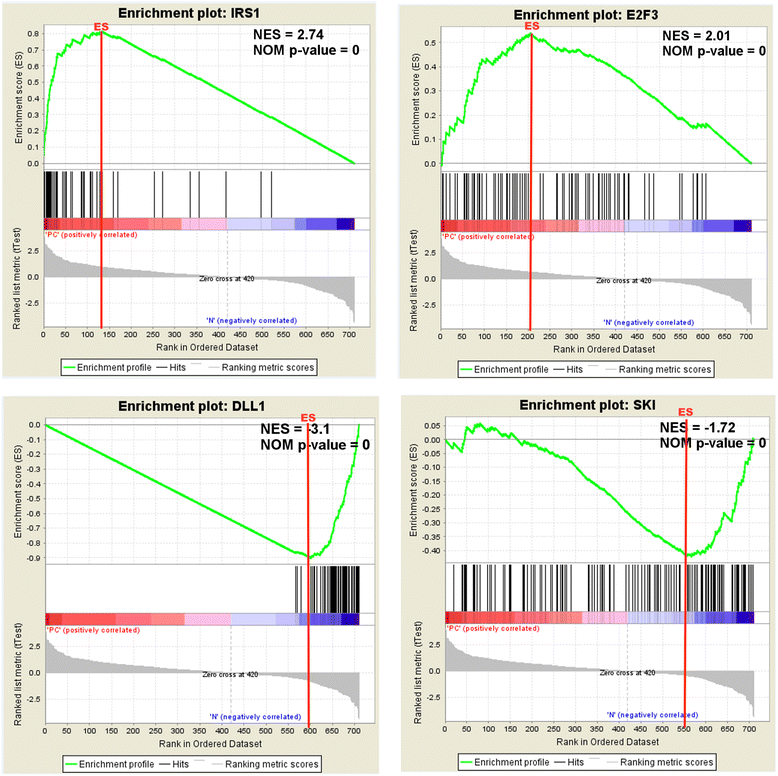
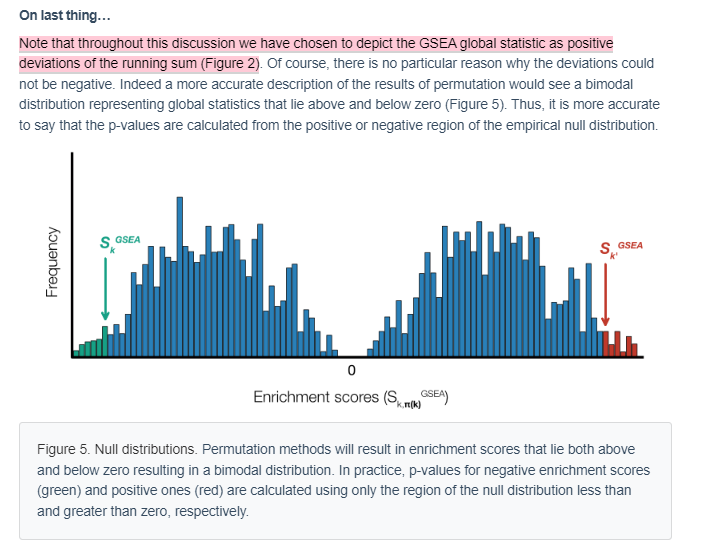
关于NES(Normalized enrichment score)
最后得到的enrichment score是和gene list有关系的,gene list越大,那么得分越高,如何去掉这部分的影响呢,normalize

关于代码部分
主要解析三部分的代码,怎么处理输入,怎么计算最大值,是怎么计算p-value
输入的处理,stats是输入的基因列表,里面主要是带有名字的score array,先排序再取绝对值,非常巧妙,那么就可以同时计算通路的上调和下调了。如果在顶端富集,就是上调,否则就是下调。
stats <- sort(stats, decreasing=TRUE)
stats <- abs(stats) ^ gseaParam
下面这一段主要是解析怎么计算最大值,稍微解读几个关键变量,S是所选pathway中基因在所有基因中的位置。r是所有基因的score,p是score在计算完绝对值之后取的值,代码主要分为两个部分,一部分在计算曲线上最高点和最低点的位置,也就是tops 和 bottoms。
scoreType <- match.arg(scoreType)
S <- selectedStats
r <- stats
p <- gseaParam
S <- sort(S)
m <- length(S)
N <- length(r)
if (m == N) {
stop("GSEA statistic is not defined when all genes are selected")
}
NR <- (sum(abs(r[S])^p))
rAdj <- abs(r[S])^p
if (NR == 0) {
# this is equivalent to rAdj being rep(eps, m)
rCumSum <- seq_along(rAdj) / length(rAdj)
} else {
rCumSum <- cumsum(rAdj) / NR
}
tops <- rCumSum - (S - seq_along(S)) / (N - m)
if (NR == 0) {
# this is equivalent to rAdj being rep(eps, m)
bottoms <- tops - 1 / m
} else {
bottoms <- tops - rAdj / NR
}
maxP <- max(tops)
minP <- min(bottoms)
一部分是在计算geneSetStatistic,这个是根据scoreType来选择我是保留正数最大值还是负数最大值还是这两个里面绝对值的最大值。
switch(scoreType,
std = geneSetStatistic <- ifelse(maxP == -minP, 0, ifelse(maxP > -minP, maxP, minP)),
pos = geneSetStatistic <- maxP,
neg = geneSetStatistic <- minP)
接下来是怎么计算p-value,可以很明显的看出是根据scoreType这个值来判断选取异常值。代码很短,一眼就能看明白。
switch(scoreType,
std = pvals[, nMoreExtreme := ifelse(ES > 0, nGeEs, nLeEs)],
pos = pvals[, nMoreExtreme := nGeEs],
neg = pvals[, nMoreExtreme := nLeEs])
关于使用场景
上面说了那么多,我什么时候用正数,什么时候用负数呢?github上面有一个issue专门提到了这个问题,我大致凝练一下,就是看你的输入,当你的输入都是正数的时候,也就是你的输入只关注这个通路有没有上调,那自然是scoreType直接用pos就可以。至于具体的更细致的原理部分,得看这篇论文Improving Gene-Set Enrichment Analysis of RNA-Seq Data with Small Replicates。作者列出的第一个原因我赞成,第二个原因我属实不理解。

后记
后续可能还需要填的坑,为什么基因的permutation因为基因内部的相关性会导致假阳性呢?自己的猜想是pathway内部基因相关性很高所以导致聚集出现从而导致假阳性。如果是sample重新排列组合,基因之间的相关性得以保留,因此不会出现假阳性,至于为什么单边实验会解决这个问题呢,不太懂。

参考文献:
1、https://www.biostars.org/p/398994/
2、https://www.pathwaycommons.org/guide/primers/data_analysis/gsea/
3、Improving Gene-Set Enrichment Analysis of RNA-Seq Data with Small Replicates
4、https://www.cnblogs.com/jiangkejie/p/11572205.html


【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· winform 绘制太阳,地球,月球 运作规律
· AI与.NET技术实操系列(五):向量存储与相似性搜索在 .NET 中的实现
· 超详细:普通电脑也行Windows部署deepseek R1训练数据并当服务器共享给他人
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 上周热点回顾(3.3-3.9)