RNA-seq | 转录组标准分析流程和常用工具软件介绍
笔记内容摘要:RNA-seq转录组基础知识与标准分析流程,简单记录学习过程。
转录组分析是对样本转录产物RNA的深入挖掘研究。通常情况下,植物的表型差异可能由许多因素控制,其中包括基因的转录环节,不同基因的转录情况有所不同,可能会使表型发生变化。
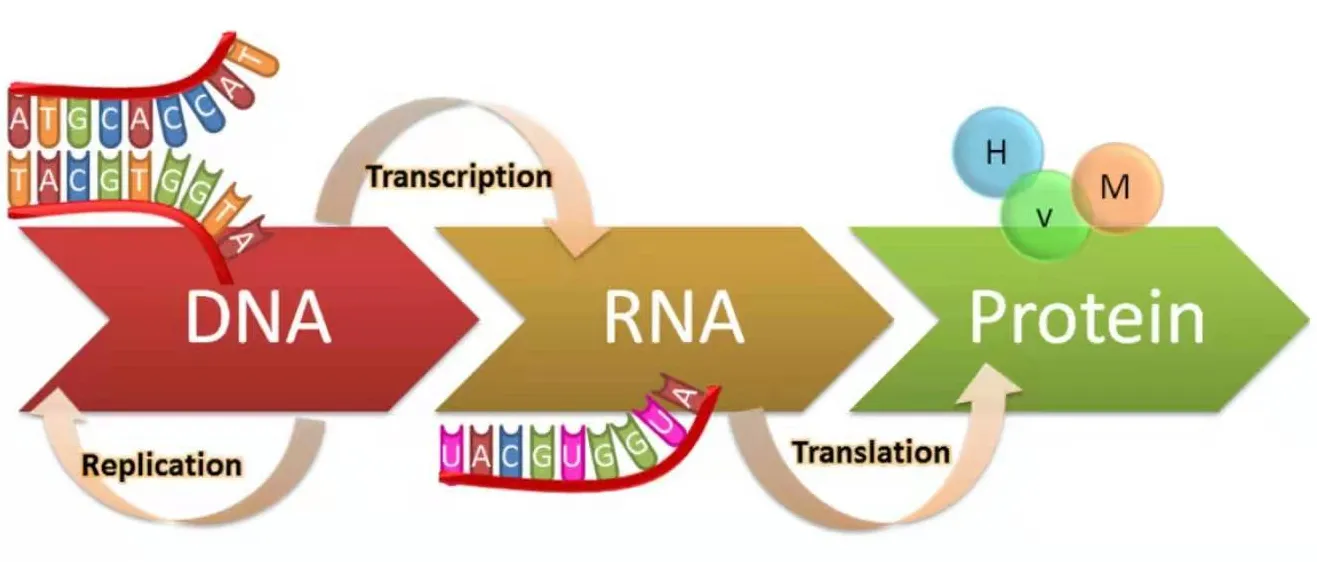
差异表达分析是对mRNA测序后获得表达矩阵,研究不同基因的表达量差异,除此之外,还有功能富集分析、联合分析等多种手段。
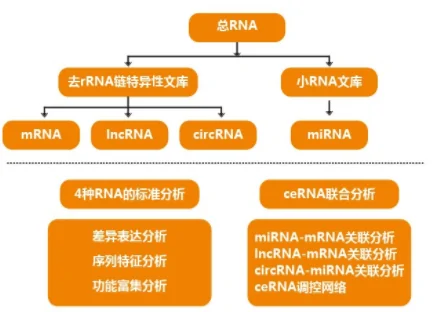
转录组分析标准流程
数据准备
- 测序数据
测序数据:fastq格式的文件(由测序公司提供),每4行为一个reads。

- 数据信息
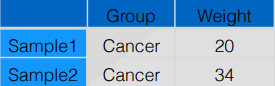
样本信息表:每行是一个样本,每列是一个性状表型。
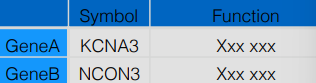
基因信息表:每行是一个基因、每列是一个信息,信息为注释所得。
- 参考基因组
1.基因组序列.fastq
2.基因注释.gtf
这一部分是前期数据准备的过程,通过测序或者数据库获取原始数据,用于后续的分析流程。
比对到参考基因组
- 操作步骤
1.构建参考基因组
2.序列比对
3.压缩和排序
4.建立索引 bam index
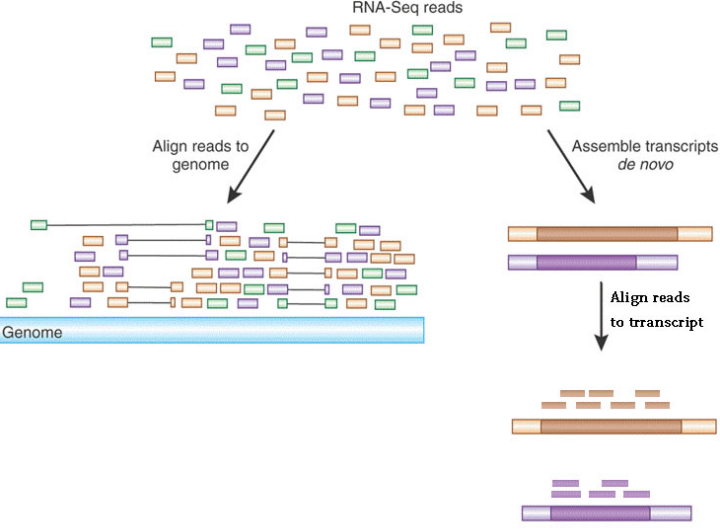
- 输出文件
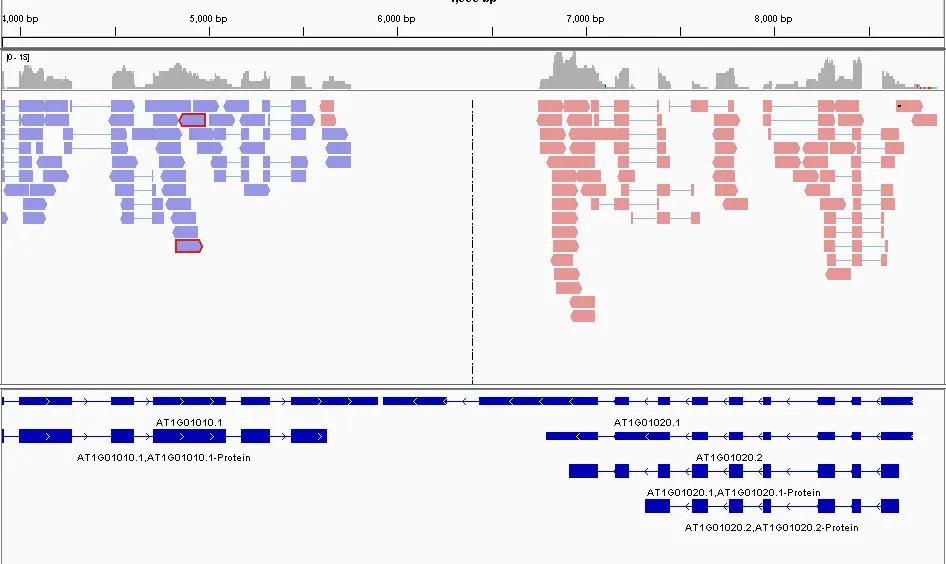
对比结果.bam利用IGV可视化对比软件能够打开bam文件进行查看。
定量表达
- 操作步骤
htseq htseq-count
subread(rsubread)
利用上述软件实现单个样本表达量的计算,另外还可以将count格式转化为FPKM格式,之后用于R语言进一步绘图使用。
- 输出文件
定量结果.count
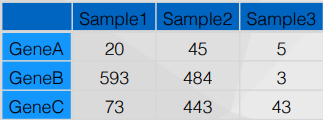
表达矩阵
表达矩阵的每一行是一个基因,每一列是一个样本。
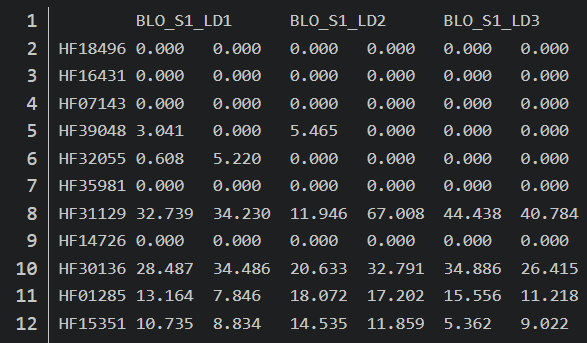
- 操作步骤
1.counts 矩阵
2.TPM 矩阵
3.TPM+TMM 矩阵
上述步骤是进行数据的标准化和处理,构建合适的表达矩阵,TPM和TMM是不同的矫正方法,形成如下的数据格式
差异表达分析
- 操作步骤
1.DESeq2
有生物学重复时使用。用于寻找组间显著表达变化的基因,DESeq2主要使用负二项分布的模型来进行差异分析。
2.edgeR
无生物学重复时使用。edgeR是一个研究重复计数数据差异表达的Bioconductor软件包。基于负二项分布的统计方法,包括经验贝叶斯估计、精确检验、广义线性模型和准似然检验。
后续还可以进行KEGG,GSEA,GO富集分析
转录组数据分析所需软件列表:
质控
fastqc , multiqc, trimmomatic, cutadapt ,trim-galore
比对
star, hisat2, bowtie2, tophat, bwa, subread
计数
htseq, bedtools, deeptools, salmon
参考资料:
https://www.genek.cn
https://blog.csdn.net/bio_meimei/article/details/109458283
https://blog.csdn.net/qq_28723681/article/details/124914014
https://blog.csdn.net/weixin_45536936/article/details/126026764
本文由mdnice多平台发布

 浙公网安备 33010602011771号
浙公网安备 33010602011771号