

1. 函数源代码
该R包中最主要的函数是 hclust ,代码如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 | > fastcluster::hclustfunction (d, method = "complete", members = NULL) { if (method == "ward") { message("The \"ward\" method has been renamed to \"ward.D\"; note new \"ward.D2\"") method <- "ward.D" } METHODS <- c("single", "complete", "average", "mcquitty", "ward.D", "centroid", "median", "ward.D2") method <- pmatch(method, METHODS) if (is.na(method)) stop("Invalid clustering method.") if (method == -1) stop("Ambiguous clustering method.") dendrogram <- c(.Call(fastcluster, attr(d, "Size"), method, d, members), list(labels = attr(d, "Labels"), method = METHODS[method], call = match.call(), dist.method = attr(d, "method"))) class(dendrogram) <- "hclust" return(dendrogram)} |
对比基础包 stats 中的函数 hclust :
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 | > stats::hclustfunction (d, method = "complete", members = NULL) { METHODS <- c("ward.D", "single", "complete", "average", "mcquitty", "median", "centroid", "ward.D2") if (method == "ward") { message("The \"ward\" method has been renamed to \"ward.D\"; note new \"ward.D2\"") method <- "ward.D" } i.meth <- pmatch(method, METHODS) if (is.na(i.meth)) stop("invalid clustering method", paste("", method)) if (i.meth == -1) stop("ambiguous clustering method", paste("", method)) n <- as.integer(attr(d, "Size")) if (is.null(n)) stop("invalid dissimilarities") if (is.na(n) || n > 65536L) stop("size cannot be NA nor exceed 65536") if (n < 2) stop("must have n >= 2 objects to cluster") len <- as.integer(n * (n - 1)/2) if (length(d) != len) (if (length(d) < len) stop else warning)("dissimilarities of improper length") if (is.null(members)) members <- rep(1, n) else if (length(members) != n) stop("invalid length of members") storage.mode(d) <- "double" hcl <- .Fortran(C_hclust, n = n, len = len, method = as.integer(i.meth), ia = integer(n), ib = integer(n), crit = double(n), members = as.double(members), nn = integer(n), disnn = double(n), diss = d) hcass <- .Fortran(C_hcass2, n = n, ia = hcl$ia, ib = hcl$ib, order = integer(n), iia = integer(n), iib = integer(n)) structure(list(merge = cbind(hcass$iia[1L:(n - 1)], hcass$iib[1L:(n - 1)]), height = hcl$crit[1L:(n - 1)], order = hcass$order, labels = attr(d, "Labels"), method = METHODS[i.meth], call = match.call(), dist.method = attr(d, "method")), class = "hclust")} |
二者的区别如下:
fastcluster::hclust 和 stats::hclust 函数都用于进行层次聚类,但它们在实现和性能上可能有差异。从 R 代码来看,它们在接口上非常相似,都需要一个距离矩阵 d 和一个指定方法 method 的参数。不过,这些函数在内部如何处理聚类过程可能有所不同。
fastcluster::hclust 是 fastcluster 包中的函数,这个包特别设计用于处理大数据集并优化性能。它提供了一个接口到 fastcluster 库,这是一个为速度优化的层次聚类算法的集合。如代码所示, fastcluster::hclust 使用 .Call 接口调用 C 语言级别的代码,这通常比 R 中的纯代码执行得更快。
stats::hclust 是 R 中 stats 包的一部分,提供了标准的层次聚类功能。代码显示它使用 .Fortran 调用 Fortran 代码来执行聚类。虽然 stats 包非常可靠并且在标准 R 安装中提供,但可能没有 fastcluster 包中的函数那么快,特别是在处理非常大的数据集时。
此外, fastcluster::hclust 在处理 "ward" 方法时,会发出警告信息指出 "ward" 方法已经更名为 "ward.D"(这里同时提到了一个新的 "ward.D2" 方法)。这表明 fastcluster 包在方法名称上可能更严格。其他方面,这两个函数的参数都很相似,例如 method , members 等。
总的来说,两者的主要区别可能在于执行速度,尤其是在大规模数据集上。 fastcluster 在算法上可能更加高效,因此对于大型数据集可能是更好的选择。对于较小的数据集,或者当性能考虑不是首要问题时, stats::hclust 或许已经足够。然而,这两个函数在 API 设计上是非常类似的,提供了相似的用户体验。在选择使用哪一个时,可能需要根据你的数据规模和性能需求来决定。
2. 代码
函数的使用代码如下
1 | sampleTree = hclust(d = dist(datExpr), method = "average") |
3. 结果解析
hclust 函数在 R 中用于层次聚类分析,其结果是一个 hclust 对象。这个对象包含进行聚类分析所需的所有信息,并能够以树状图(dendrogram)的形式展现。
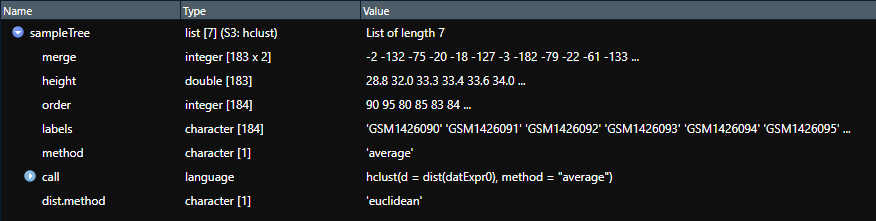
下面是 hclust 结果对象的组成部分和它们各自的含义:
1. merge:一个矩阵,描述了在聚类过程中各个聚类是如何合并的。每一行代表一个合并的操作,如果值为负数,表示它指向单个初始观测点;如果为正数,表示它指向由 merge 矩阵之前行的合并结果形成的聚类。
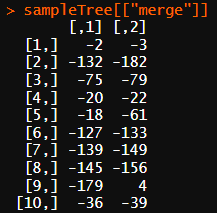
2. height:一个向量,包含了在聚类树中每次合并发生时的“高度”。这个“高度”可以根据所选的相异度测量方法(比如,最大距离、平均距离、Ward's 方法等)而有所不同。
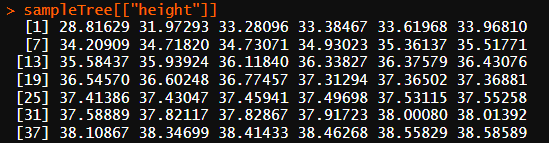
3. order:一个向量,表示了叶子节点在绘制树状图时的顺序,可以用来帮助在二维平面上更好地展示树状图。
4. labels:一个字符向量,包含了观测点的标签。在树状图中,每个叶子节点旁边通常会显示对应的标签。
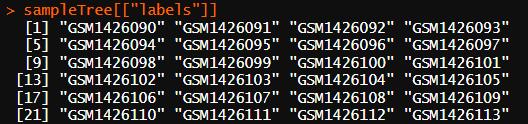
5. method:表示用于聚类的方法,比如 "complete" 、 "average" 、 "single" 等。
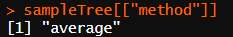
6. call:一个R语言的调用对象,表示了 hclust 函数的原始调用方式。

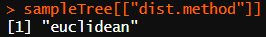
7. dist.method:当使用 dist 函数计算相异度矩阵时使用的方法。
4. 聚类结果可视化
一个 hclust 对象通常通过 plot 函数来可视化,这会生成一棵树状图,反映了数据点是如何层次性地聚类在一起的。
例如:
1 2 3 4 5 6 | # 计算距离矩阵dist_matrix <- dist(mydata) # 进行层次聚类hc <- hclust(dist_matrix) # 绘制树状图plot(hc) |
在上面的代码中, mydata 是原始数据集, dist_matrix 是计算出的距离矩阵。 hclust 函数使用这个距离矩阵执行层次聚类,并返回聚类结果 hc 。然后,我们可以通过 plot(hc) 来绘制树状图。
5. 聚类结果分组
cutree 函数有助于我们通过指定聚类数 k 来切割树状图,从而得到聚类的分组结果。也可以使用 cutreeStatic 函数对树状图进行切割。
cutree 函数和 cutreeStatic 函数的区别如下:
cutree 和 cutreeStatic 都是 R 语言中的函数,用于在层次聚类树(dendrogram)中切割分支,从而将数据分成不同的簇或者组。他们的功能是相似的,但有一些关键的区别:
1. cutree:
cutree 是 R 的基础统计包中的函数。这是一个普遍用于层次聚类分析的标准函数。它可以根据指定的簇的数量 k 或者指定的高度 h 来切割聚类树。当基于簇的数量来切割时, cutree 会尝试保持簇的数量尽可能接近你指定的 k 值。但是由于它没有内置的最小簇大小限制,因此可能会生成一些非常小的簇。
举个例子:
1 2 3 | clusters <- cutree(hc, k = 5)# 或者clusters <- cutree(hc, h = 1.25) |
在这个例子中, hc 是之前通过 hclust 得到的层次聚类对象, k = 5 指示 cutree 函数将数据点分成5个簇。 h = 1.25 导致树状图在高度1.25处被切割,从而将观测点分配到不同的簇中。结果是,所有在高度1.25以下相连的点都会被划分为同一个簇,而高度在1.25以上的点会分属于不同的簇。
2. cutreeStatic:
cutreeStatic 是专门设计用于 WGCNA 包中的,它也执行恒定高度的切割,但使用了额外的条件限制生成的簇。
cutreeStatic 允许用户指定一个最小簇大小( minSize ),这有助于避免划分出太小的簇,这在基因表达数据分析中是有意义的,因为太小的模块可能在统计学上不够稳健。 cutreeStatic 返回的每个簇至少有 minSize 个成员,否则这些对象会被标记为0(未分配)。
使用 cutreeStatic 的例子:
1 2 3 | # 假设 dendro 是通过 hclust 得到的层次聚类树状图library(WGCNA)clusters <- cutreeStatic(dendro, cutHeight = 0.9, minSize = 50) |
在 cutreeStatic 中, cutHeight 参数定义了树应该在何处高度上切割, minSize 参数则确保只有足够大的簇才被保留。相比之下, cutree 可以返回任意大小的簇。
总结来说, cutree 更适用于一般的层次聚类和簇的快速提取,而 cutreeStatic 提供了更多控制,特别是在需要避免过小的簇时,这在 WGCNA 的上下文中尤其有价值。
6. 分组结果解析
cutreeStatic 函数的输出是一个数值向量,它给出了层次聚类树状图上每个对象的簇标签。在这个向量中,具有相同数值的对象被归为同一个簇,而数值为0的对象表示未被分配到任何簇。这通常意味着树在指定的高度上被切割,然后剔除了那些成员数量少于 `minSize` 参数指定值的分支。
举例来说,如果你有一个包含100个对象的数据集,并执行了 cutreeStatic 函数,得到了如下类型的输出:
1 | [1] 1 1 1 2 2 2 0 0 3 3 3 3 3 0 4 4 4 4 4 4 0 0 5 5 ... |
在这个例子中,前三个对象(值为1)被归为第一个簇,接着的三个对象(值为2)形成第二个簇。值为0的对象没有被分配到任何簇,可能因为这些对象所属的原始分支在切割时没有足够的成员数满足 minSize 的要求。随后的五个对象(值为3)构成了第三个簇,如此类推。
cutreeStatic 函数会根据簇的大小来赋予簇编号,其中最大的簇会被标记为1,第二大的簇标记为2,以此类推。这样的编号方式有助于快速识别数据集中最大的几个簇,这在许多情况下很有用,尤其是在生物信息学和网络分析中,大的簇可能代表了更有生物学意义的模块。
需要注意的是, cutreeStatic 特别适用于 WGCNA 分析,因为在该框架中,研究者通常对较大的簇或模块更感兴趣,这些簇可能代表了具有共同生物学功能的基因集合。小簇可能是噪声或者不具有足够统计力的信号,因此在某些情况下被排除在外是合理的。


【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 全程不用写代码,我用AI程序员写了一个飞机大战
· DeepSeek 开源周回顾「GitHub 热点速览」
· 记一次.NET内存居高不下排查解决与启示
· 物流快递公司核心技术能力-地址解析分单基础技术分享
· .NET 10首个预览版发布:重大改进与新特性概览!
2023-02-16 生信分析
2023-02-16 TCGA代码分析流程 - 3.2 生存分析:log-rank,Cox回归