

正则化、R包glmnet包:https://www.cnblogs.com/xiaogaobugao/p/17149331.html
LASSO回归
什么是Lasso回归
Tibshirani(1996) 引入了 LASSO (Least Absolute Shrinkage and Selection Operator)模型,用于参数的选择和收缩。当我们分析大数据时,这个模型非常有用。可以用R包 glmnet 包建立 LASSO 模型。
Lasso回归是一种线性回归模型,也被称为正则化或惩罚回归模型,它使用L1正则化项来限制变量的系数。这意味着 Lasso 回归可以用来选择最重要的变量,并且可以让不重要的变量的系数收缩到0。因此 Lasso 可以用于变量数量较多的大数据集。而传统的线性回归模型无法处理这类大数据。
Lasso回归复杂度调整的程度由参数lambda来控制,lambda越大模型复杂度的惩罚力度越大,从而获得一个较少变量的模型。Lasso回归和bridge回归都是Elastic Net广义线性模型的特例。除了参数lambda,还有参数alpha,控制对高相关性数据时建模的形状。Lasso回归,alpha=1(R语言glmnet的默认值),brigde回归,alpha=0,一般的elastic net 0<alpha<1.
为了选择最重要的变量,Lasso 回归使用了贪心算法。它会从所有变量中选择一个变量,然后计算使用这个变量时模型的误差。然后,它会继续从剩余的变量中选择一个变量,并计算使用这个变量时模型的误差。这个过程会不断重复,知道找到最优的变量子集为止。
虽然线性回归估计器 (linear regression estimator)在偏-方差权衡关系方面是无偏估计器,但正则化或惩罚回归,如Lasso,Ridge承认一些减少方差的偏倚。这意味着后者的最小化问题有两个组成部分:均方误差(linear regression estimator)和惩罚参数。Lasso 的L1惩罚使变量选择和收缩成为可能,而Ridge的L2惩罚使变量收缩成为可能。

由上式可知,Lasso 回归公式的第一部分是残差平方和(Residual Sum of Squares,RSS),第二部分是惩罚项。该罚项由超参数 λ 调整,λ 由用户通过人工搜索或交叉验证的方式外源性给出。
当Lasso 中包含了某些变量,但RSS值的降低很小,可以忽略不计时,收缩惩罚的影响就会增加。这意味着这个变量的系数是零(Lasso)或接近零(Ridge)。
Lasso回归使用L1正则化项来限制系数,可以帮助减少变量数量,避免模型过度拟合,并提高模型的泛化能力。
Lasso回归与岭回归的区别
Lasso回归和岭回归都是常用的正则化技术,他们都可以用来减少系数的大小,但他们使用的正则项不同。Lasso回归使用L1正则项,岭回归使用L2正则项。L1正则项的特点是,它可以完全移除某些系数,使得模型中的变量更加稀疏,即变量较少。这对于特征选择非常有用。而岭回归不具有变量的筛选功能,它只能让变量之间的差异变得更小。但L1正则项也有一些缺点,由于L1正则项对所有变量的惩罚都是绝对值,所以对于变量之间的大小没有影响。因此,L1正则项会使得变量之间存在较大的差异,导致模型的可解释性降低。
Lasso回归的优点
Lasso回归的优点在于能够选择出最重要的特征,并且能够有效地防止过拟合。在实际应用中,Lasso回归常用于预测和特征选择。
Lasso回归的缺点
Lasso回归的缺点在于当特征之间存在线性关系时,它的表现不够优秀,以及当特征的数量较多时,计算目标函数的值会变得困难。
比起岭回归,Lasso所带的L1正则项对于系数的惩罚要重得多,并且它会将系数压缩至0,由此选择那些系数不为0的那些特征,以供后续线性回归建模使用。Lasso的正则化系数λ系数更加敏感,因此将其控制在很小的空间范围内变动,以此来寻找最佳的正则化系数。
相比于岭回归,Lasso可以解决现在高维数据一个普遍问题——稀疏性。高维数据即n>m的情况,现在随着数据采集能力的提高,特征数采集的多,但是其中可能有很多特征是不重要的,实际系数很小,如果用岭回归可能估计出相反的结果,即系数很大。而用Lasso可以把这些不重要变量的系数压缩为0,既实现了较为准确的参数估计,也实现了特征选择即降维。
与岭回归(Ridge Regression)相比,Lasso回归亦可解决多重共线性问题,但是不一样的是Lasso回归针对不同的自变量,会使其收敛的速度不一样。有的变量就很快趋于0了,有的却会很慢。因此一定程度上Lasso回归非常适合于做特征选择。但Lasso不是从根本上解决多重共线性问题,而是限制多重共线性带来的影响。
asso回归在建立广义线型模型的时候,可以包含一维连续因变量、多维连续因变量、非负次数因变量、二元离散因变量、多元离散因变,除此之外,无论因变量是连续的还是离散的,lasso都能处理,总的来说,lasso对于数据的要求是极其低的,所以应用程度较广。
lasso的复杂程度由λ来控制,λ越大对变量较多的线性模型的惩罚力度就越大,从而最终获得一个变量较少的模型。
1. 准备输入数据
rm(list = ls())
library(glmnet) # 正则化技术
library(car) # 多重共线性检验
library(corrplot) # 相关性绘图
library(survival) # 生存分析
library(dplyr) # 数据处理
library(ggpubr) #基于ggplot2的可视化包ggpubr
library(patchwork) # 拼图
library(ROCR) # 画ROC曲线
library(caret) # 切割数据
library(ggplot2) # 高级画图必备
setwd("D:/BioInformationAnalyze/TCGAdata\CRC")
load("TCGA-COAD_04.lncRNA_SurvModel_LassoData.Rdata")
输入数据格式如下:

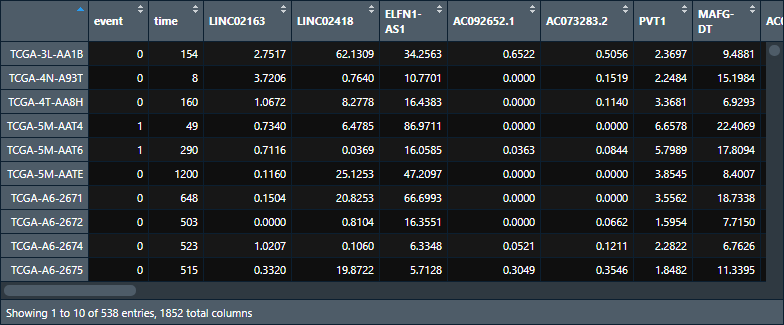
2. 数据检查
# 数据检查 class(datExpr) # 检查输入数据的格式。目前,glmnet包只能接受矩阵形式的数据,数据框的数据会报错,所以我们先要把数据转换成矩阵形式,这一步很重要。 summary(datExpr$time) # 检查时间的单位 table(datExpr$event) # 检查生存状态 min(datExpr$time) # 查看最小值,看是否有0 datExpr <- filter(datExpr, time > 0) # 删除数据框中time值小于0的样本,有0的话后续分析会报错,而且这些样本的随访时间为0,没有生存分析的意义 data_use <- datExpr # 数据备份 nrow(data_use) # 查看样本数 ncol(data_use) # 查看变量数
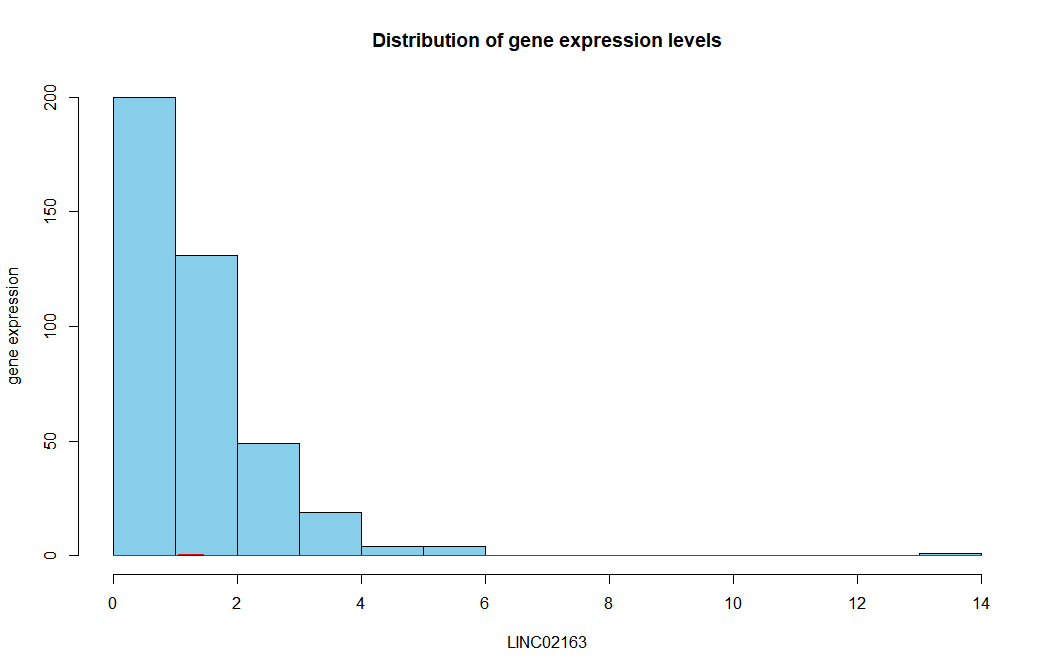
(1)绘制直方图和正态分布概率密度函数图,查看数据分布
# 绘制直方图和正态分布概率密度函数图
hist(data_use$LINC02163, main = "Distribution of gene expression levels",
xlab = "LINC02163", ylab = "gene expression", col = "skyblue") # 分布直方图
curve(dnorm(x, mean = mean(data_use$LINC02163), sd = sd(data_use$LINC02163)), add = TRUE, col = "red") # 正态分布的概率密度函数
# shapiro-wilk检验
shapiro.test(data_use$LINC02163) # 适用于小样本数据,N≤50,若p值<0.05,则数据不符合正态分布
# Kolmogorov-Smirnov检验(柯尔莫哥洛夫检验)
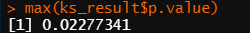
ks.test(data_use$LINC02163, pnorm, mean = mean(data_use$LINC02163), sd = sd(data_use$LINC02163)) # 适用于大样本数据,N>50,若p值<0.05,则数据不符合正态分布
# 对所有列的数据进行Kolmogorov-Smirnov循环检验
ks_result <- data.frame("Gene" = character(), "p.value" = numeric())
for (i in 3:length(colnames(data_use))){
p.value <- ks.test(data_use[,i], pnorm, mean = mean(data_use[,i]), sd = sd(data_use[,i]))$p.value
Gene <- colnames(data_use)[i]
tmp <- data.frame(Gene = Gene,
p.value = p.value)
ks_result <- rbind(ks_result, tmp)
}
max(ks_result$p.value) # 若p值<0.05,则数据不符合正态分布
直方图正态分布概率密度函数图:
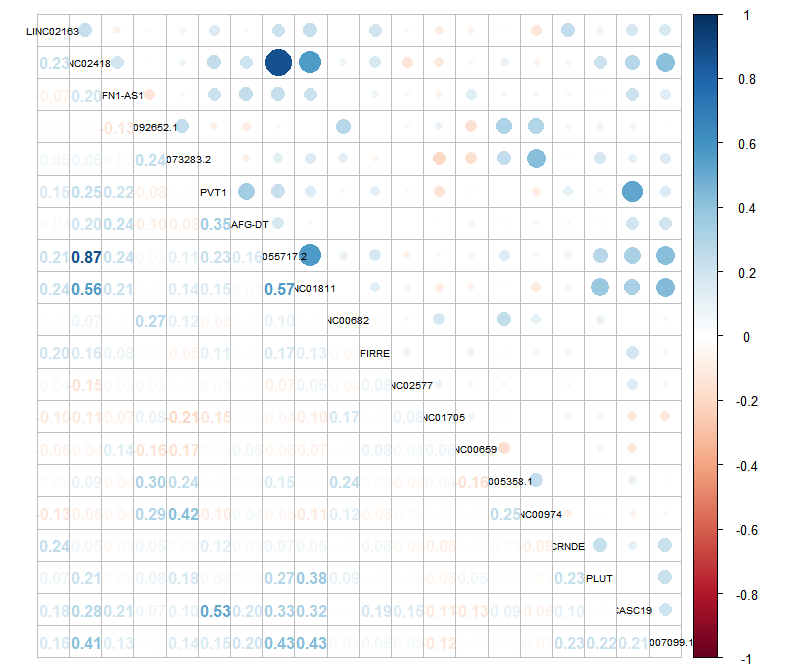
(2)检查变量之间的相关性,绘制相关性热图,检查数据有无多重共线性问题
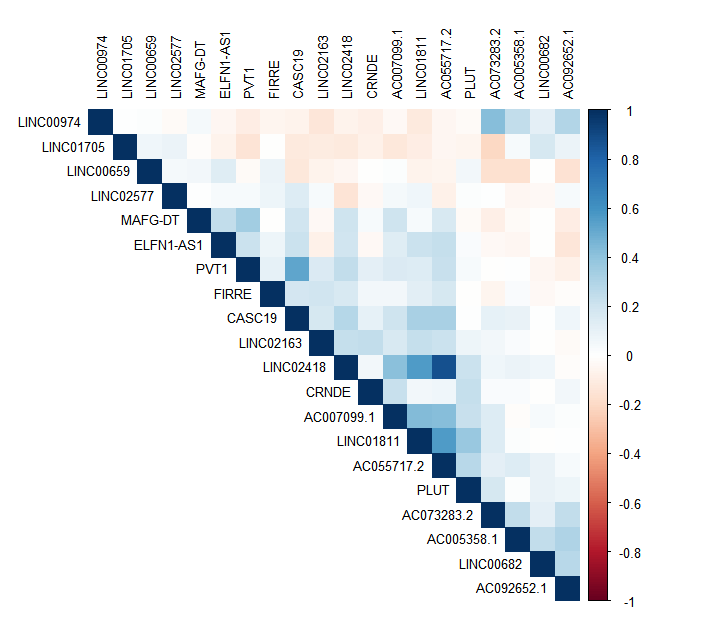
# 绘制相关性热图,检查数据的多重共线性问题
cor_matrix <- cor(data_use[,3:22], use = "everything", method = c("spearman"))
corrplot(cor_matrix, type = "upper", method = "color", tl.cex = 0.8, tl.col = "black", order = "AOE")
corrplot.mixed(cor_matrix, tl.cex = 0.6, tl.col = "black") # 如果相关性不大于0.8,则认为这份数据的多重共线性问题没有那么严重。
相关性热图,检查多重共线性:
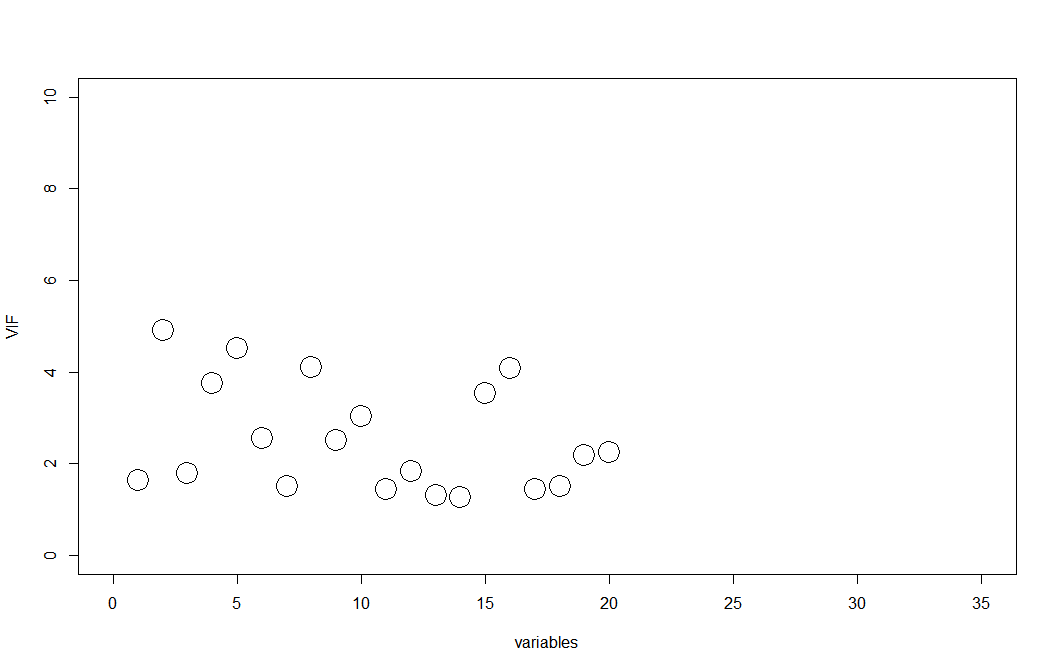
(3)多重共线性检查 之 方差膨胀因子(VIF)
# 多重共线性检查之方差膨胀因子(VIF)
data_use1 <- data_use[,1:22]
model <- coxph(Surv(time, event) ~ ., data = data_use1)
vif(model)
plot(vif(model),
xlim = c(0,35), ylim = c(0,10),
xlab = "variables", ylab = "VIF",
cex = 3, pch = 1)
# 当VIF大于5时,认为存在多重共线性,当VIF大于10时,认为存在严重的多重共线性
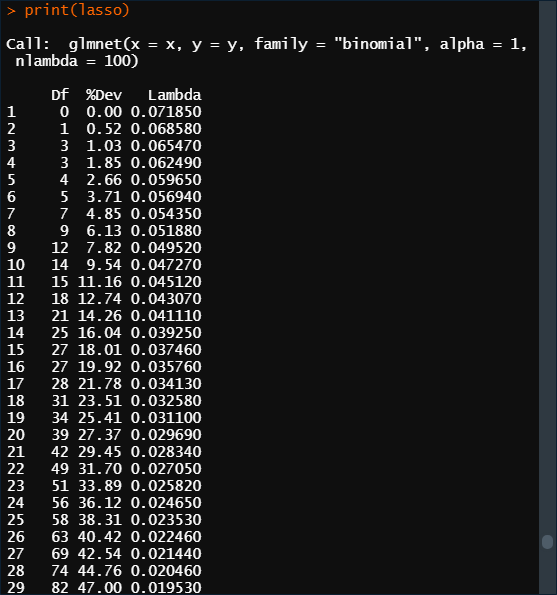
2. 构建Lasso回归模型
time <- data_use[,"time"]
status <- data_use[,"event"]
x <- as.matrix(data_use[,-c(1,2)]) # x为输入特征,应该是矩阵格式。否则数据框的数据会报错:Error in lognet...(串列)对象不能强制改变成'double'种类
y <- as.matrix(data_use$event))
lasso <- glmnet(x = x, y = y,
family = "binomial",
alpha = 1, # alpha = 1为LASSO回归,= 0为岭回归,0和1之间则为弹性网络
nlambda = 100) # nlambda表示正则化路径中的个数,这个参数就可以起到一个阈值的作用,决定有多少基因的系数可以留下来。默认值为100。
print(lasso)
……
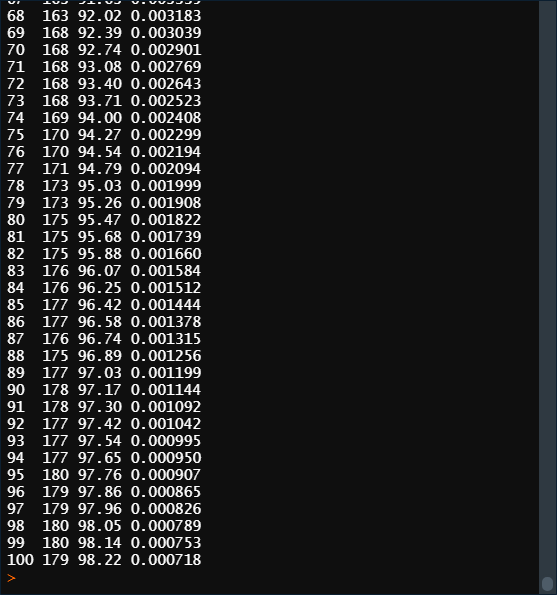
解释一下输出结果三列的意思:
Df:自由度,表示使用的变量数量,
%Dev:代表了由模型解释的残差的比例。对于线性模型来说就是模型拟合的R^2(R-squred)。它在0%和100%之间,越接近100%说明模型包括了越多样本的信息,表现越好,如果是0%,说明模型的预测结果还不如直接把因变量的均值作为预测值来的有效。解释的残差百分比越高越好,但是构建模型使用的基因的数量也不能太多,需要取一个折中值。
Lambda:表示使用的正则化系数,是构建模型的重要参数。数量和nlambda的值一致。
plot(lasso, xvar = "lambda", label = TRUE) # 系数分布图,由“log-lambda”与变量系数作图,展示根据lambda的变化情况每一个特征的系数变化,展示了Lasso回归筛选变量的动态过程 plot(lasso, xvar = "dev", label = TRUE) # 也可以对%dev绘图 plot(lasso, xvar = "norm", label = TRUE) # “L1范数”与变量系数作图
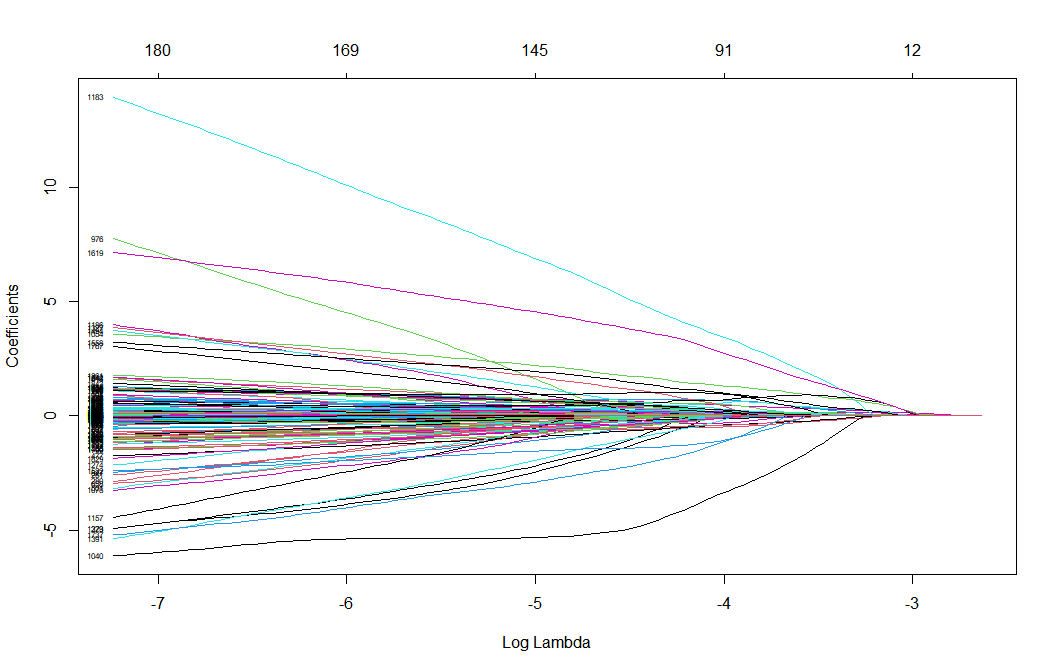
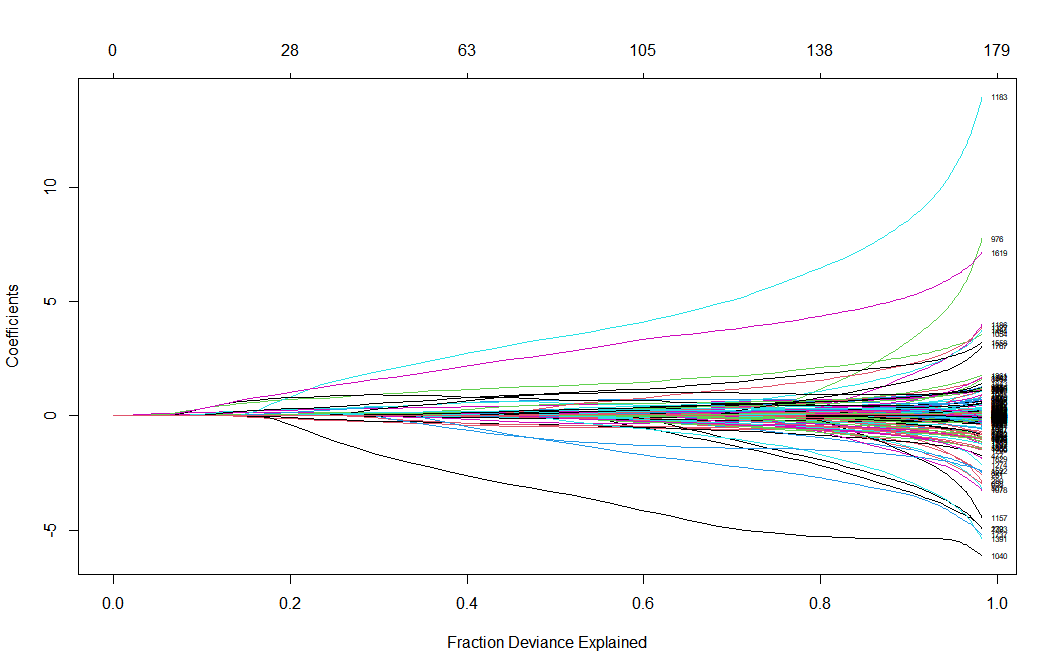
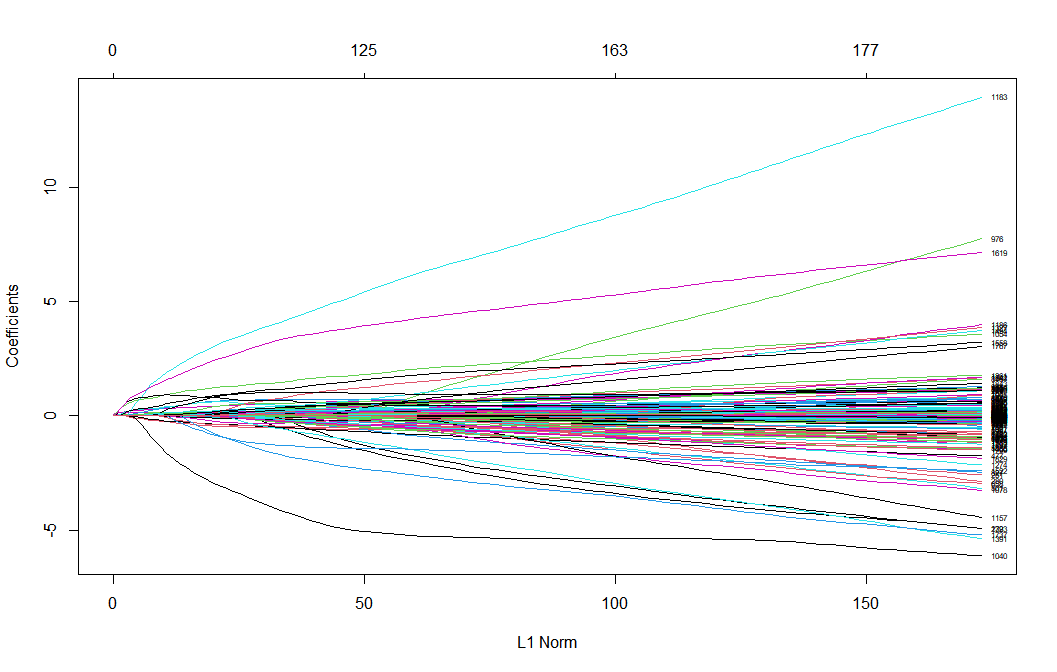
在系数分布图中,下横坐标为lambda的对数,上横坐标为当前的变量数,纵坐标为变量的系数。一条线就代表一个基因,这些基因的末尾会指向一个纵坐标,这个纵坐标就是系数,LASSO会为每一个基因算一个系数。但这些基因的系数并不是都能用于后续分析的,这就需要一个参数λ,这个参数λ就可以起到一个阈值的作用,决定有多少基因的系数可以留下来。可以看到随着lambda的增加,变量系数不断减少,部分变量系数变为0(等于没有这个变量了)。
在回归方程中,回归系数可以理解为自变量对因变量影响大小的参数,当回归系数归零,则变量被剔除出方程
coef_lasso <- coef(lasso, s = 0.000718) # 查看每个变量的回归系数。s为lasso回归的结果中的lambda值,选取相应的λ值可以得到相应数量的变量的回归系数 coef_lasso
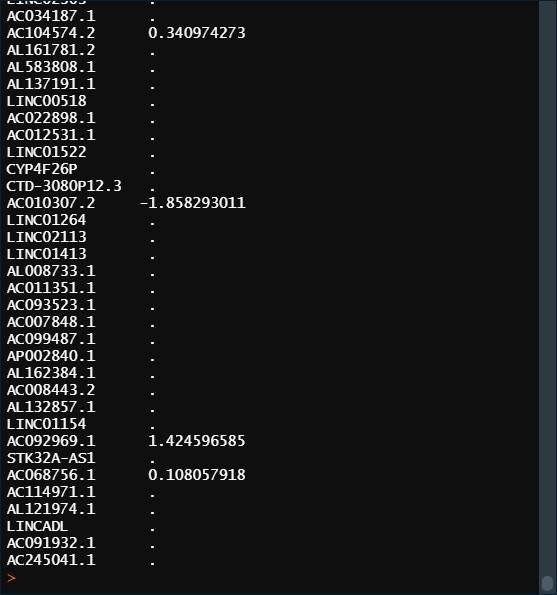
3. 交叉验证,挑选合适的λ值,变量筛选
模型已经跑出来了,筛选变量还得确定lambda,也就是在上图中画一条垂直于横轴的直线,这样我们才能知道哪些变量被压缩为0,以及未被压缩为0的变量的系数的估计值究竟是多少。
可以使用R的glmnet::cv.glmnet。这个函数使用的是“交叉验证”挑选lambda。type.measure表示模型的损失函数,默认为均方误差,nfolds指定k的大小,默认是20折交叉验证。
set.seed(1234567) # 设置种子数,保证每次得到的交叉验证图是一致的 # 计算100个λ,画图,筛选表现最好的λ值 lasso_cv <- cv.glmnet(x = x, y = y, family = "binomial", alpha = 1, nlambda = 100) # 交叉验证,如果结果不理想,可以重新单独运行这一行代码,或者换一下种子数 plot(lasso_cv) # 纵坐标:部分似然偏差。上横坐标:当前的变量数。下横坐标:log(λ)
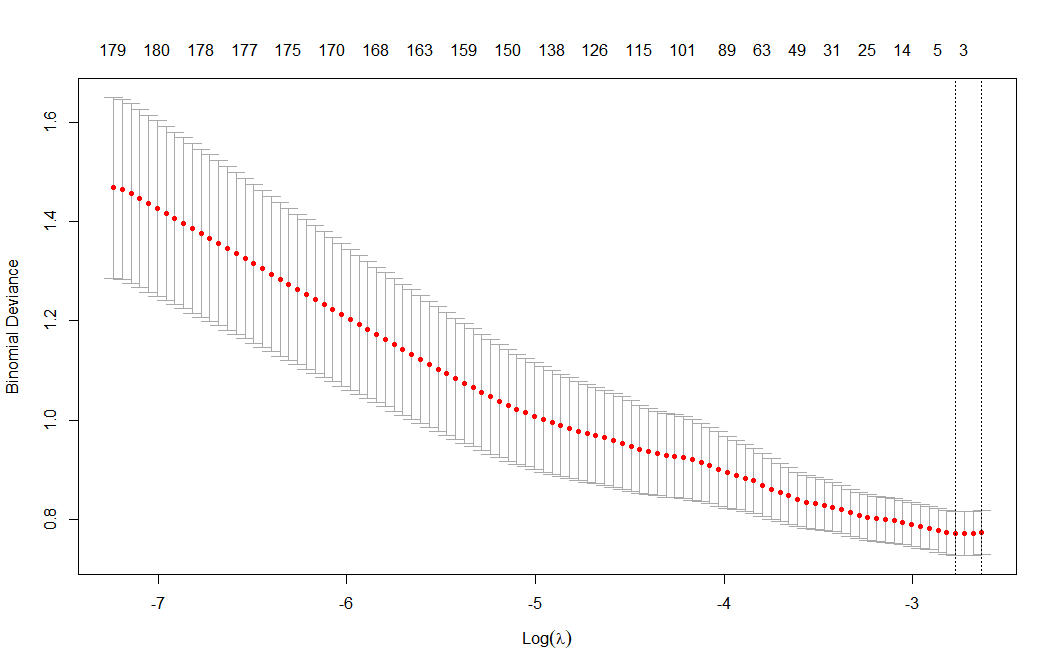
这张图上有两条虚线,这两条虚线分别指示了两个特殊的λ值: 左边是lambda.min(平均误差最小时的λ值),右边是lambda.1se(平均误差在一个标准差以内的最大λ值)。这两个值之间的lambda都认为是合适的。lambda.1se构建的模型最简单,即使用的基因数量少,而lambda.min则准确率更高一点,使用的基因数量更多一点。一般会选择lambda.min。
虚线上方对应的数字表示有多少个基因的系数可以留下来。具体的系数和基因还是要看LASSO回归的结果变量。这张图及上面的系数分布图其实主要还是展示一下大体的结果。
lambda <- lasso_cv$lambda.min # lambda <- lasso_cv$lambda.1se lambda coef_lasso_cv <- coef(lasso, s = lambda) # 查看每个变量的回归系数。s为lasso回归结果中的lambda值,可以得到相应数量的变量的回归系数 coef_lasso_cv[,1][coef_lasso_cv[,1]!=0]
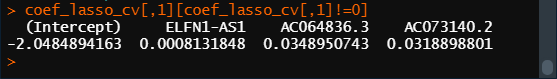
exp(coef_lasso_cv[,1][coef_lasso_cv[,1]!=0]) # 自然对数的指数函数,它的定义为exp(x) = e^x,其中e是自然对数的底数(约等于2.718),回归系数<0的exp>1,反之<1。
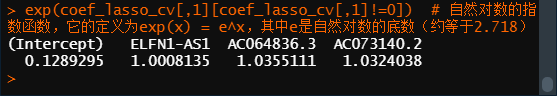
4.用这两个λ值重新建模
model_lasso_min <- glmnet(x = x, y = y, alpha = 1, lambda = lasso_cv$lambda.min) # model_lasso_1se <- glmnet(x = x, y = y, alpha = 1, lambda = lasso_cv$lambda.1se) # 这两个值体现在参数lambda上。有了模型,可以将筛选的基因挑出来了。 # 所有基因存放于model_lasso_min模型的子集beta中,用到的基因有一个s0值,没用的基因只记录了“.”,所以可以用下面代码挑出用到的基因。 model_lasso_min$beta[,1][model_lasso_min$beta[,1]!=0] choose_gene_min = rownames(model_lasso_min$beta)[as.numeric(model_lasso_min$beta) != 0] # choose_gene_1se = rownames(model_lasso_1se$beta)[as.numeric(model_lasso_1se$beta) != 0] length(choose_gene_min) # length(choose_gene_1se)
model_lasso_min:
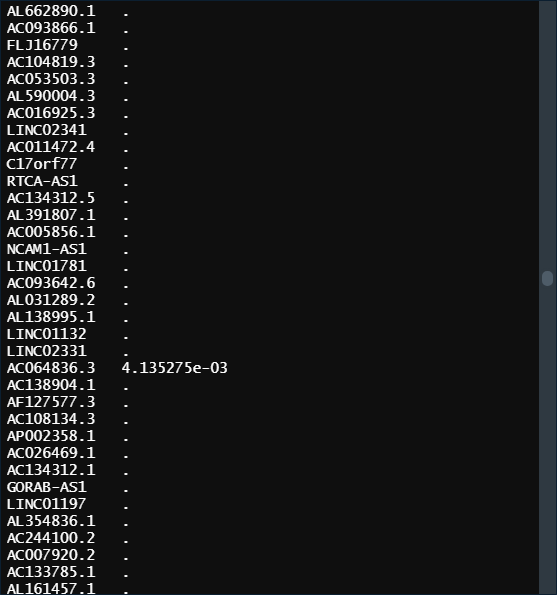
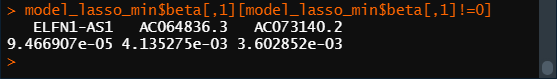

如变量没有显示系数,即lasso回归收缩系数为零。这意味着它完全被排除在模型之外,因为它的影响力不够。系数非0的变量即为我们筛选的重要特征。
这样我们就得到了最终的lasso回归模型以及各个参数的回归系数。
5. 模型预测和评估 - 自己预测自己
可以使用最终的Lasso回归模型对新的观测进行预测。这里先预测一下自己本身,看从自己身上得到的模型能否对自己做出较好的预测。
(1)模型预测
lasso.prob <- predict(lasso_cv, newx = x, s = c(lasso_cv$lambda.min, lasso_cv$lambda.1se))
输出结果lasso.prob是一个矩阵:第一列是min的预测结果,第二列是1se的预测结果。预测结果是概率,或者说百分比,不是绝对的0和1。
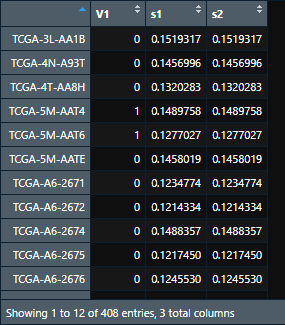
re = cbind(y, lasso.prob) # 将每个样本的生死和预测结果放在一起,直接cbind即可。 head(re)
(2)箱线图(预测结果可视化)
# 对预测结果进行可视化。以实际的生死作为分组,画箱线图整体上查看预测结果。
re = as.data.frame(re)
colnames(re) = c("event", "prob_min", "prob_1se") # 改列名
re$event = as.factor(re$event) # 画图时需要factor,分别看lasso模型对生、死两组病人的预测情况。
library(ggpubr)
p1 = ggboxplot(re, x = "event", y = "prob_min", # x轴为生死,y轴为lambda.min模型的预测值
color = "event", palette = "jco",
add = "jitter") +
scale_y_continuous(limits = c(0,1)) + # 调整y轴范围
stat_compare_means() # 将均值比较的p值添加到ggplot
p2 = ggboxplot(re, x = "event", y = "prob_1se", # x轴为生死,y轴为lambda.1se模型的预测值
color = "event", palette = "jco",
add = "jitter") +
scale_y_continuous(limits = c(0,1)) +
stat_compare_means()
library(patchwork)
p1 + p2
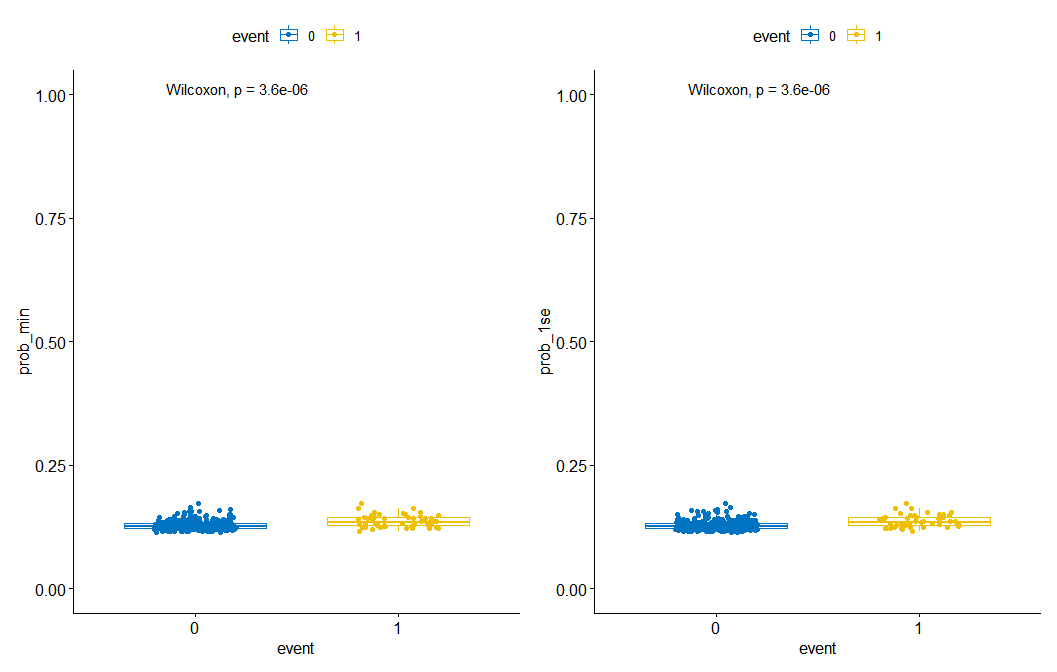
可以看到,真实结果是生存(0)的样本,预测的值就小一点(靠近0),真实结果是死亡(1)的样本,预测的值就大一点(靠近1),整体上趋势是对的,但不是完全准确,模型是可用的。对比两个λ值构建的模型,差别不大,min的预测值准确一点。
该箱线图是为了比较两个模型哪个预测结果更好,不放进论文中。
(3)ROC曲线(模型评估)
ROC曲线全称为受试者工作特征曲线 (receiver operating characteristic curve),它是根据一系列不同的二分类方式(分界值或决定阈),以真阳性率(敏感性)为纵坐标,假阳性率(1-特异性)为横坐标绘制的曲线。
AUC(Area Under Curve)被定义为ROC曲线下与坐标轴围成的面积,AUC就是衡量学习器优劣的一种性能指标。从定义可知,AUC可通过对ROC曲线下各部分的面积求和而得。显然这个面积的数值不会大于1。又由于ROC曲线一般都处于 y=x 这条直线的上方,所以AUC的取值范围在0.5和1之间。AUC越接近1.0,检测方法真实性越高;等于0.5时,则真实性最低,无应用价值。
library(ROCR)
library(caret)
# 自己预测自己
# min
pred_min <- prediction(re[,2], re[,1]) # 用预测值匹配真实值,计算预测值的准确率。将输入数据转换为标准化格式。
auc_min = performance(pred_min, "auc")@y.values[[1]] # 计算AUC(ROC 曲线下的面积)
perf_min <- performance(pred_min, "tpr", "fpr") # 计算True positive rate(真阳性率,敏感性)和False positive rate(假阳性率,1-特异性)
plot(perf_min, colorize = FALSE, col = "blue") # 用"tpr"和"fpr"画ROC曲线
lines(c(0,1), c(0,1), col = "gray", lty = 4) # 给ROC曲线加上参考线
text(0.8, 0.2, labels = paste0("AUC = ", round(auc_min, 3))) # 给ROC曲线加上AUC值,保留3位小数
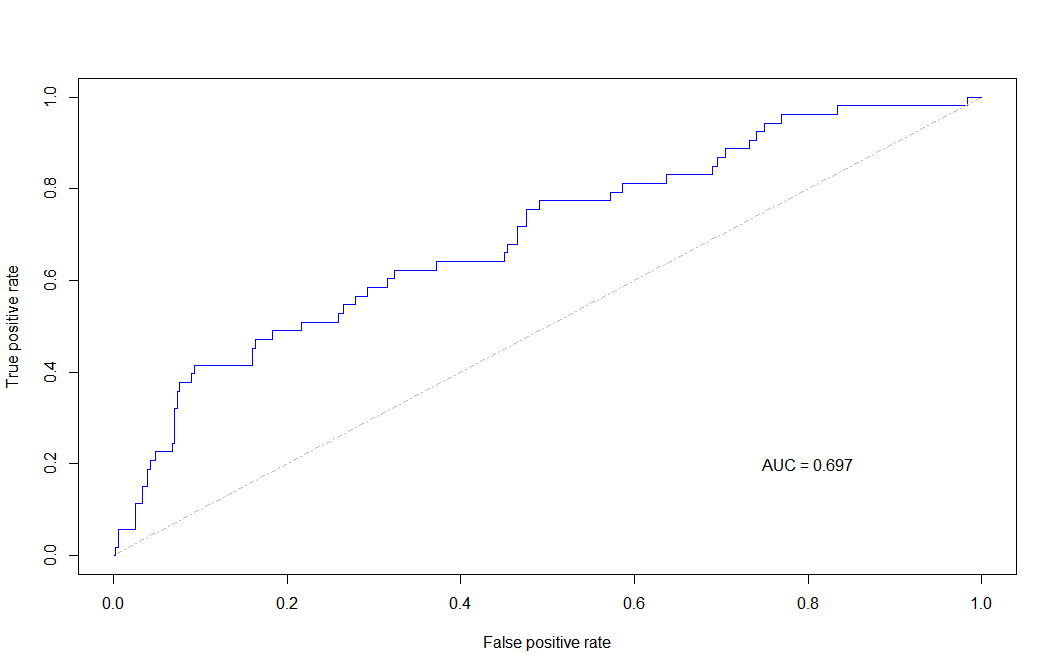
# 1se
pred_1se <- prediction(re[,3], re[,1]) # 同上
auc_1se = performance(pred_1se, "auc")@y.values[[1]] # 同上
perf_1se <- performance(pred_1se, "tpr", "fpr") # 同上
plot(perf_1se, colorize = FALSE, col = "red") # 同上
lines(c(0,1), c(0,1), col = "gray", lty = 4) # 同上
text(0.8, 0.2, labels = paste0("AUC = ", round(auc_1se, 3))) # 同上
由于交叉验证中的lambda_1se预测出来没有变量,所以得到的预测结果1se和min一样了
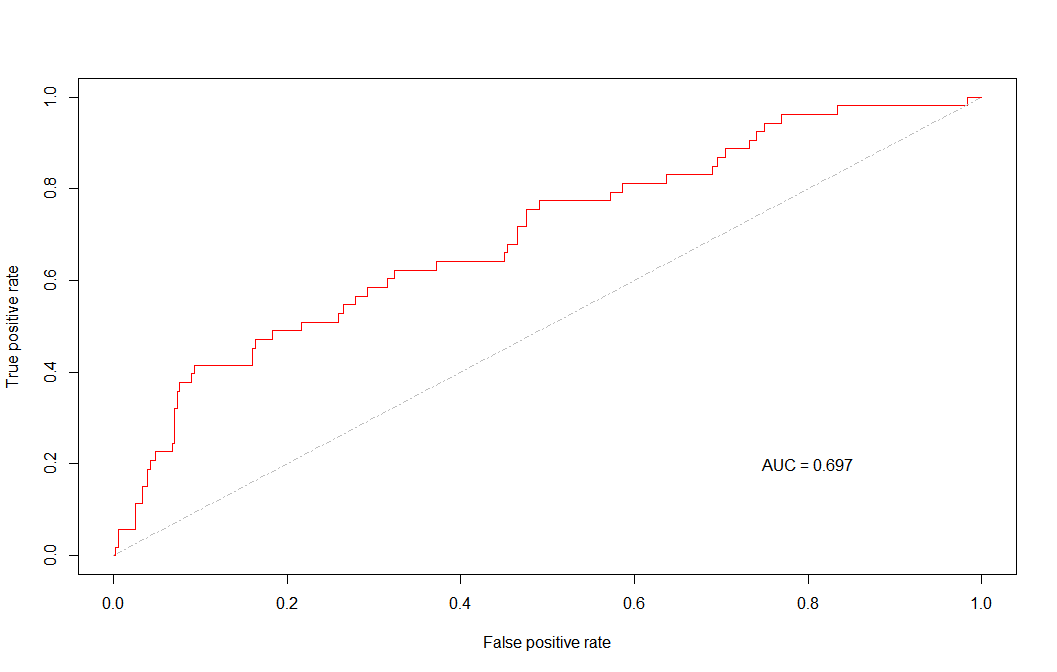
如果min和1se不一样的话,可以画在一张图上看差别:
# 把两个模型画一起
plot(perf_min, colorize = FALSE, col = "blue")
plot(perf_1se, colorize = FALSE, col = "red", add = T)
lines(c(0,1),c(0,1), col = "gray", lty = 4 )
text(0.8,0.3, labels = paste0("AUC = ", round(auc_min,3)), col = "blue")
text(0.8,0.2, labels = paste0("AUC = ", round(auc_1se,3)), col = "red")

# 用ggplot2画的更好看一点
dat = data.frame(tpr_min = perf_min@y.values[[1]],
fpr_min = perf_min@x.values[[1]],
tpr_1se = perf_1se@y.values[[1]],
fpr_1se = perf_1se@x.values[[1]])
library(ggplot2)
# min
ggplot() +
geom_line(data = dat,aes(x = fpr_min, y = tpr_min), color = "blue") + # # 用"fpr"和"tpr"画ROC曲线
geom_line(aes(x = c(0,1), y=c(0,1)), color = "grey") + # 给ROC曲线加上参考线
theme_bw() + # 去掉灰色背景
annotate("text",
x = 0.75, y = 0.25,
label = paste("AUC of min = ", round(auc_min, 2)),
color = "blue") + # 给ROC曲线加上AUC值,保留2位小数
scale_x_continuous(name = "fpr") + # x轴比例范围
scale_y_continuous(name = "tpr") # y轴比例范围
# 把两个模型画一起
ggplot() +
geom_line(data = dat,aes(x = fpr_min, y = tpr_min), color = "blue") +
geom_line(data = dat,aes(x = fpr_1se, y = tpr_1se), color = "red") +
geom_line(aes(x = c(0,1), y=c(0,1)), color = "grey") +
theme_bw() +
annotate("text",
x = 0.75, y = 0.25,
label = paste("AUC of min = ", round(auc_min, 2)),
color = "blue") +
annotate("text",
x = 0.75, y = 0.15,
label = paste("AUC of 1se = ", round(auc_1se, 2)),
color = "red") +
scale_x_continuous(name = "fpr") +
scale_y_continuous(name = "tpr")
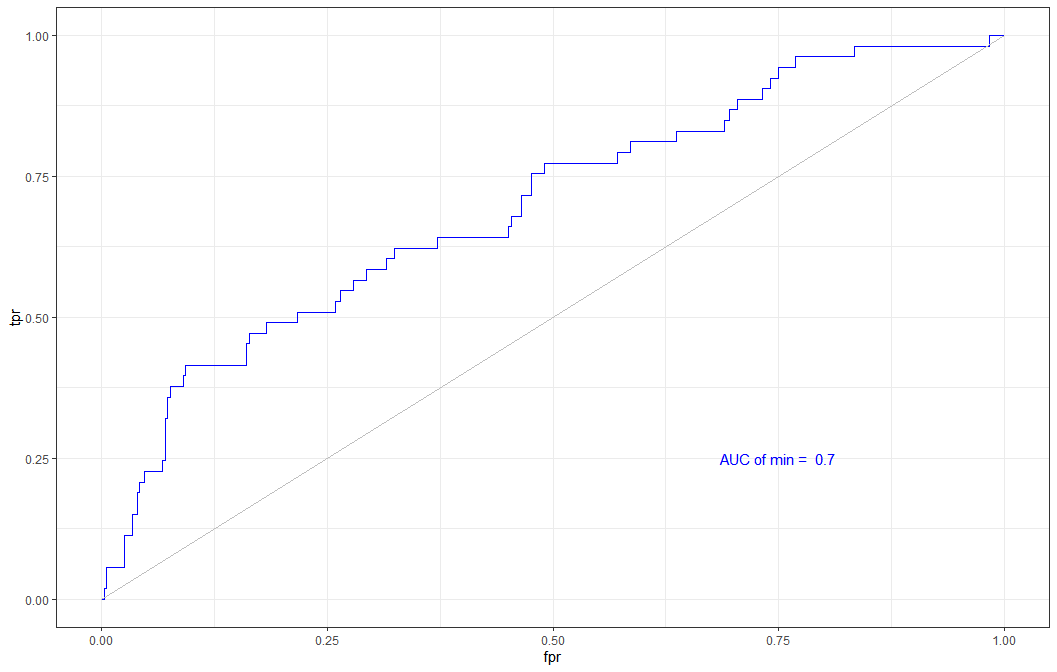
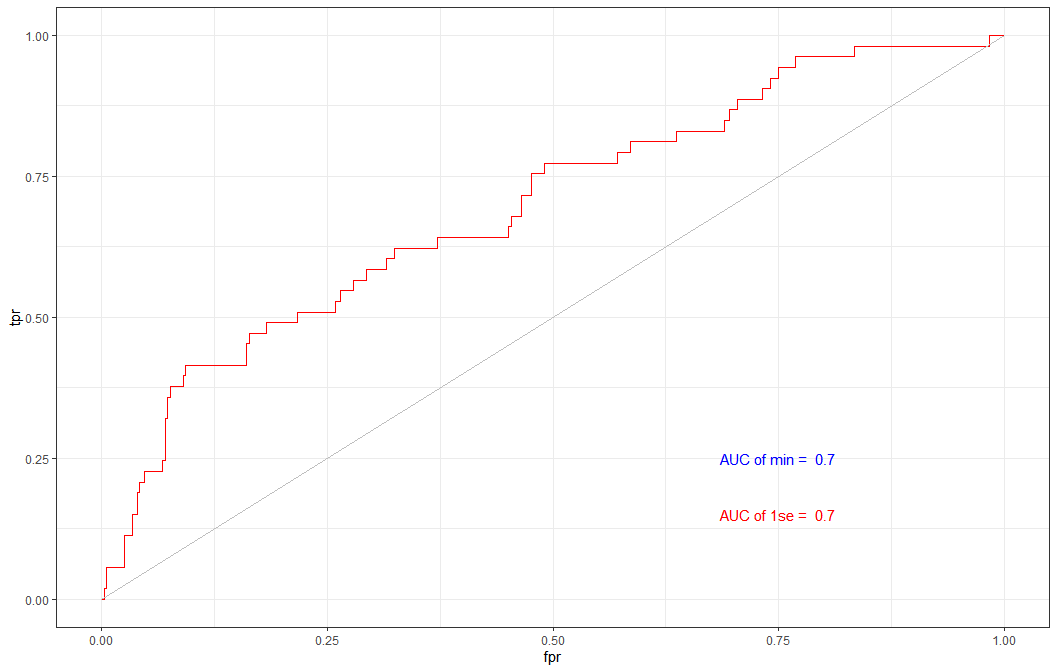
6. 模型预测和评估 - 切割数据构建模型并预测
(1)切割数据
用R包 caret 切割数据,生成的结果是一组代表列数的数字,用这些数字来给表达矩阵和 meta 取子集即可。
library(caret) set.seed(12345679) # 设置随机种子,可修改,每个种子可将数据集分成固定的训练集和测试集 sam <- createDataPartition(data_use$event, p = 0.5, list = FALSE) # createDataPartition()函数:将数据集拆分为训练集和测试集。p:用于训练集的数据百分比 head(sam) # 可查看两组一些临床参数切割比例: train <- cbind(data_use[, 1:2], data_use[, -c(1,2)][sam, ]) test <- cbind(data_use[, 1:2], data_use[, -c(1,2)][-sam, ]) prop.table(table(train$event)) prop.table(table(test$event))
(2)切割后的train数据集建模
和之前的建模方法一致
#计算lambda x = as.matrix(train[,-c(1,2)]) y = as.matrix(train$event) cv_fit <- cv.glmnet(x = x, y = y, family = "binomial", nlambda = 100, alpha = 1) plot(cv_fit) #构建模型 model_lasso_min <- glmnet(x = x, y = y, family = "binomial", alpha = 1, lambda=cv_fit$lambda.min) # model_lasso_1se <- glmnet(x = x, y = y, family = "binomial", alpha = 1, lambda=cv_fit$lambda.1se) #挑出基因 head(model_lasso_min$beta) choose_gene_min = rownames(model_lasso_min$beta)[as.numeric(model_lasso_min$beta) != 0] # choose_gene_1se = rownames(model_lasso_1se$beta)[as.numeric(model_lasso_1se$beta) != 0] length(choose_gene_min) # length(choose_gene_1se)
(3)模型预测
# 用训练集构建模型,预测测试集的生死,注意newx参数变了。
lasso.prob <- predict(cv_fit, newx = as.matrix(test[,-c(1,2)]),
s = c(cv_fit$lambda.min, cv_fit$lambda.1se))
re = cbind(event = test$event, lasso.prob)
head(re)
(4)再画箱线图(预测结果可视化)
re = as.data.frame(re)
colnames(re) = c("event", "prob_min", "prob_1se")
re$event = as.factor(re$event)
library(ggpubr)
p1 = ggboxplot(re, x = "event", y = "prob_min",
color = "event", palette = "jco",
add = "jitter") +
scale_y_continuous(limits = c(0,1)) +
stat_compare_means()
p2 = ggboxplot(re, x = "event", y = "prob_1se",
color = "event", palette = "jco",
add = "jitter") +
scale_y_continuous(limits = c(0,1)) +
stat_compare_means()
library(patchwork)
p1 + p2
(5)再画ROC曲线(模型评估)
library(ROCR)
library(caret)
# 训练集模型预测测试集
# min
pred_min <- prediction(re[,2], re[,1])
auc_min = performance(pred_min, "auc")@y.values[[1]]
perf_min <- performance(pred_min, "tpr", "fpr")
# 1se
pred_1se <- prediction(re[,3], re[,1])
auc_1se = performance(pred_1se, "auc")@y.values[[1]]
perf_1se <- performance(pred_1se, "tpr", "fpr")
# 画ROC曲线数据
dat = data.frame(tpr_min = perf_min@y.values[[1]],
fpr_min = perf_min@x.values[[1]],
tpr_1se = perf_1se@y.values[[1]],
fpr_1se = perf_1se@x.values[[1]])
# 画ROC曲线
ggplot() +
geom_line(data = dat,aes(x = fpr_min, y = tpr_min),color = "blue") +
geom_line(data = dat,aes(x = fpr_1se, y = tpr_1se),color = "red")+
geom_line(aes(x=c(0,1),y=c(0,1)),color = "grey")+
theme_bw()+
annotate("text",x = .75, y = .25,
label = paste("AUC of min = ",round(auc_min,2)),color = "blue")+
annotate("text",x = .75, y = .15,label = paste("AUC of 1se = ",round(auc_1se,2)),color = "red")+
scale_x_continuous(name = "fpr")+
scale_y_continuous(name = "tpr")
# 图中可以发现,AUC值比不拆分时降低。
用的数据不是太好,导致结果不符合预期。后续用其他数据做出好结果时再修改图片。

