
GEO背景知识 + 分析思路
实验设计
实验目的:通过基因表达量数据的差异分析和富集分析来解释生物学现象
病变组织 vs 健康组织
药物处理 vs 对照组
......
有差异的材料 → 差异基因 → 代谢通路 / 功能注释 → 解释差异的原理
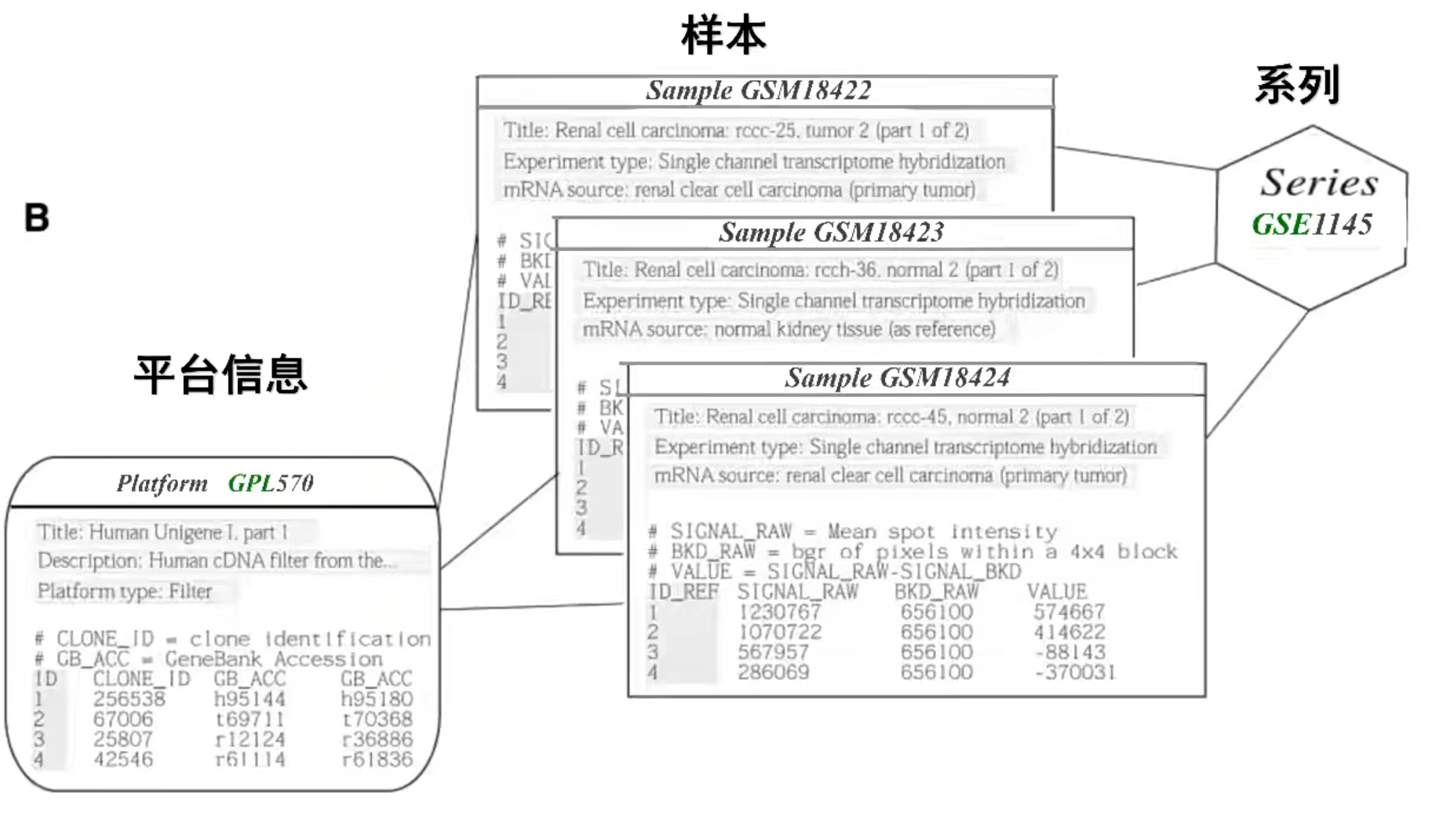
GEO数据:
用户提交给GEO的样本数据。(GSM)
一个完整的研究,并提供了整个研究的描述,包括对数据的描述,总结分析。(GSE)
用户测序使用的芯片 / 平台。(GPL)
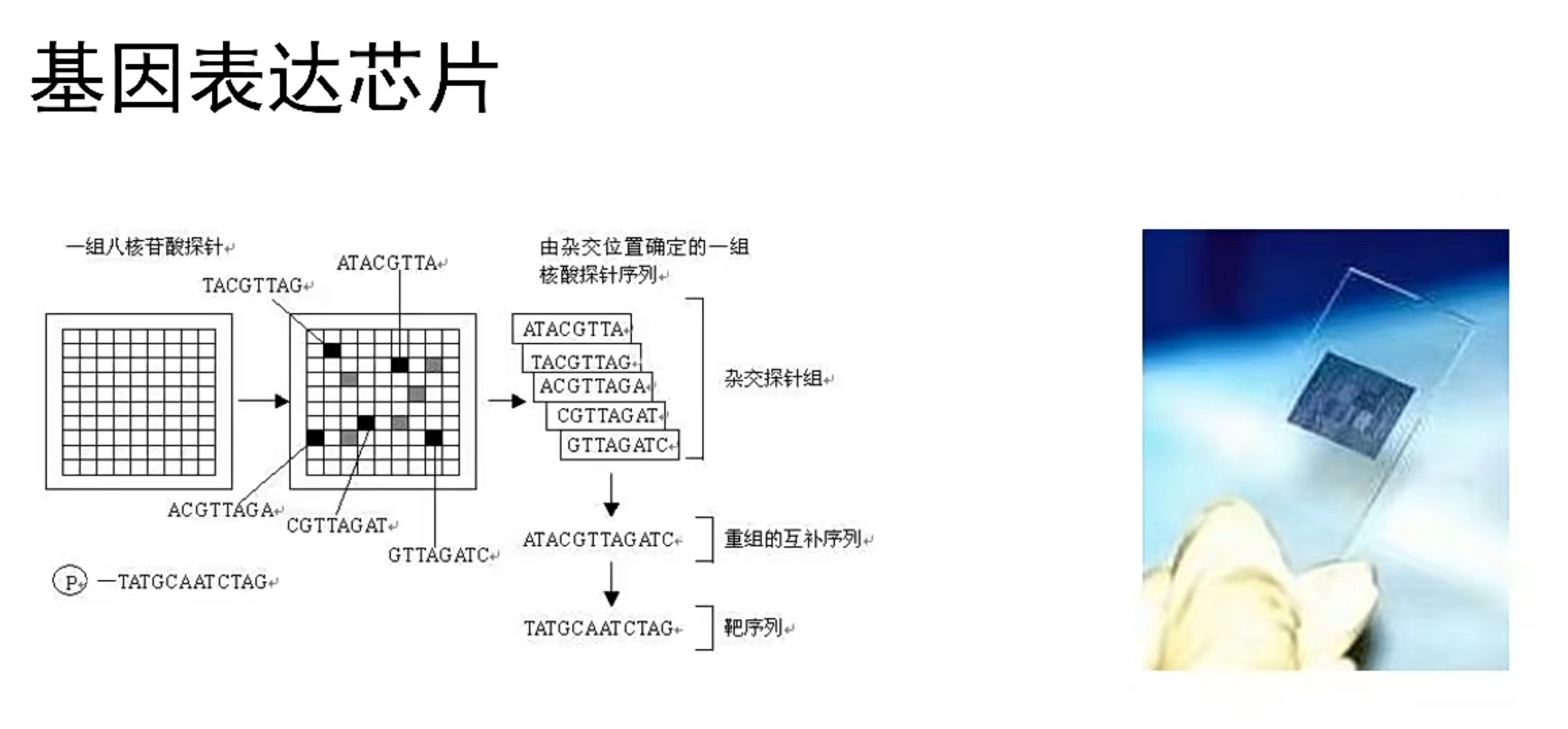
基因表达芯片的原理:探针的表达量反应基因的表达量。
分析思路
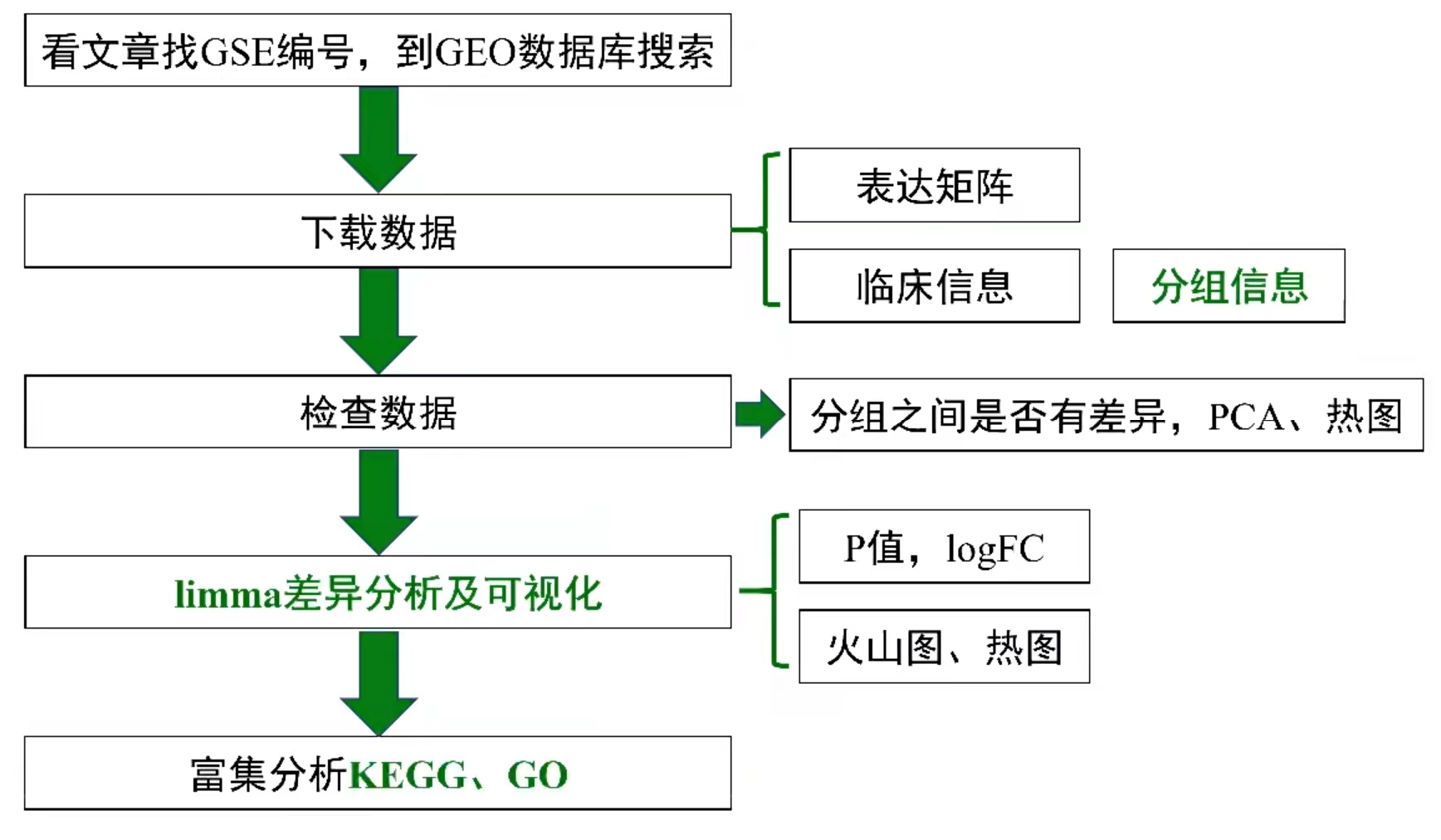
1. 看文章找GSE编号,到GEO数据库搜索
2. 下载数据:表达矩阵、临床信息
1. 将数据下载到本地
下载用函数实现:
GEOquery::getGEO
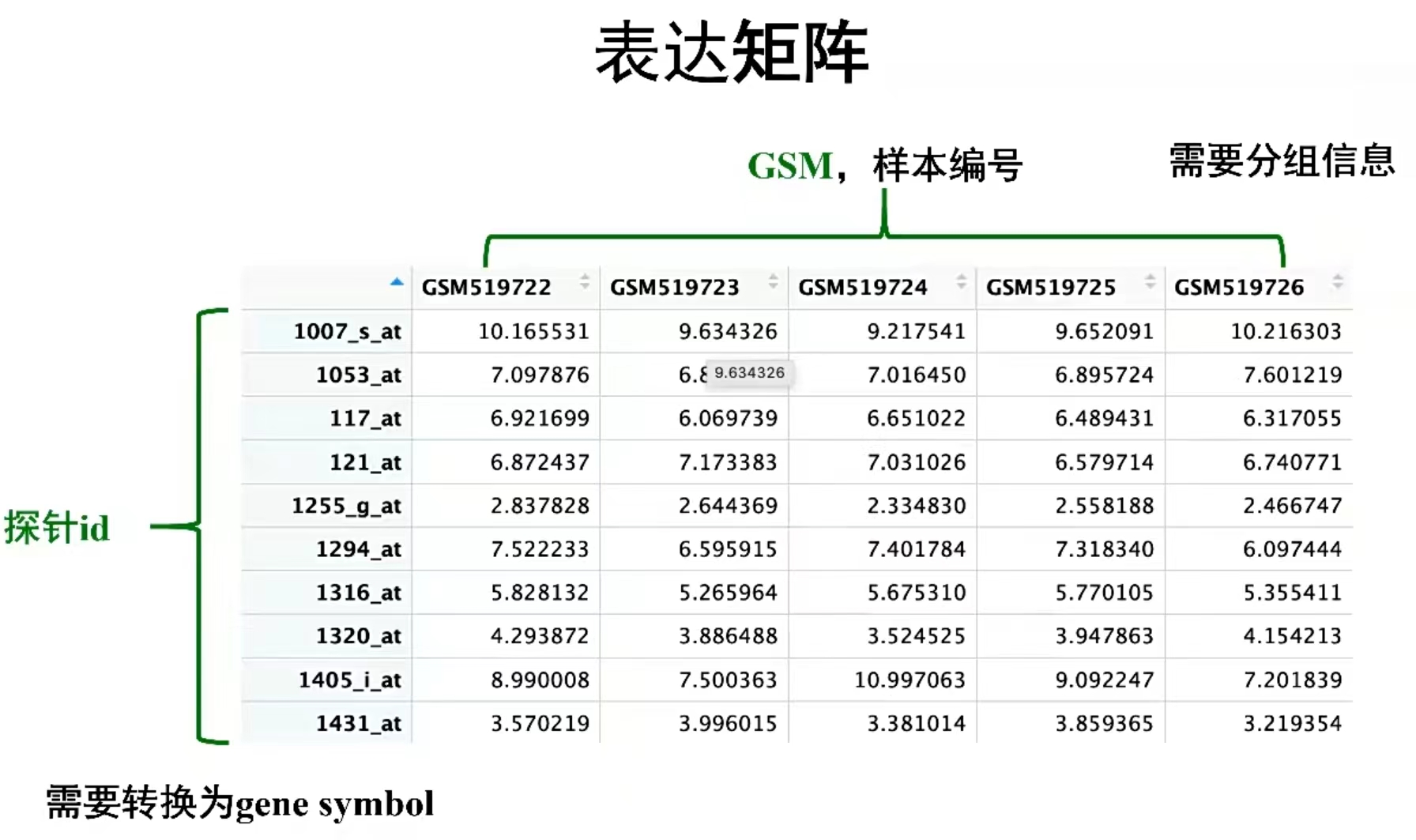
2. 以对象的形式读入R(eSet)
-从eSet中提取表达矩阵 exp(做分析的主体数据)
-从eSet中提取临床信息 pd(数据框)(提取分组信息)
-从eSet中提取 gpl 编号(寻找探针和基因之间对应关系的依据)
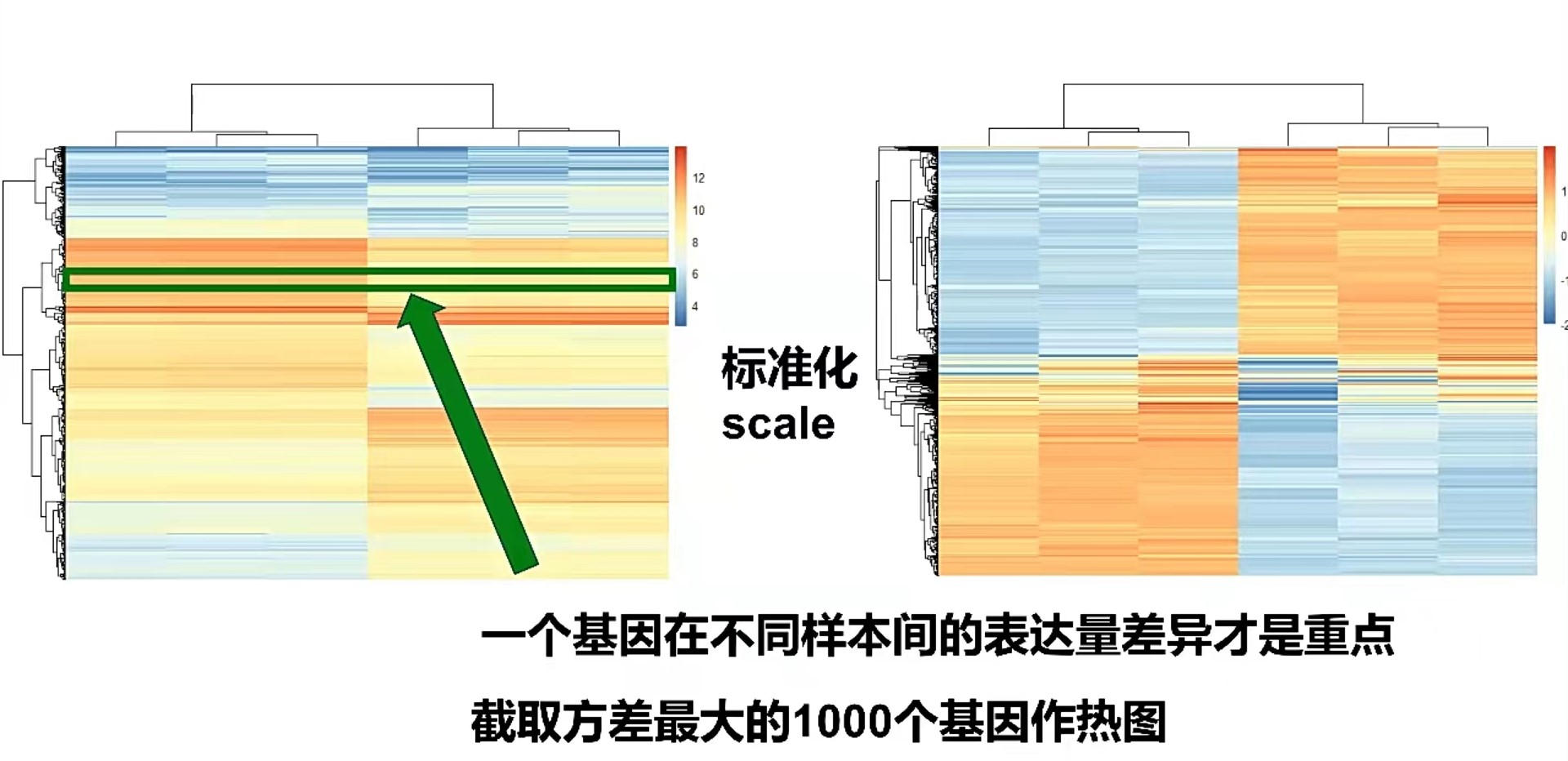
3. 检查数据:分组之间是否有差异,PCA,热图
数据框转置:
as.data.frame(t(exp))
热图标准化:
scale不需要比较同一样本中的两个基因的绝对数值大小,一个基因在不同样本间的表达量差异才是重点。
截取方差最大的1000个基因作热图:
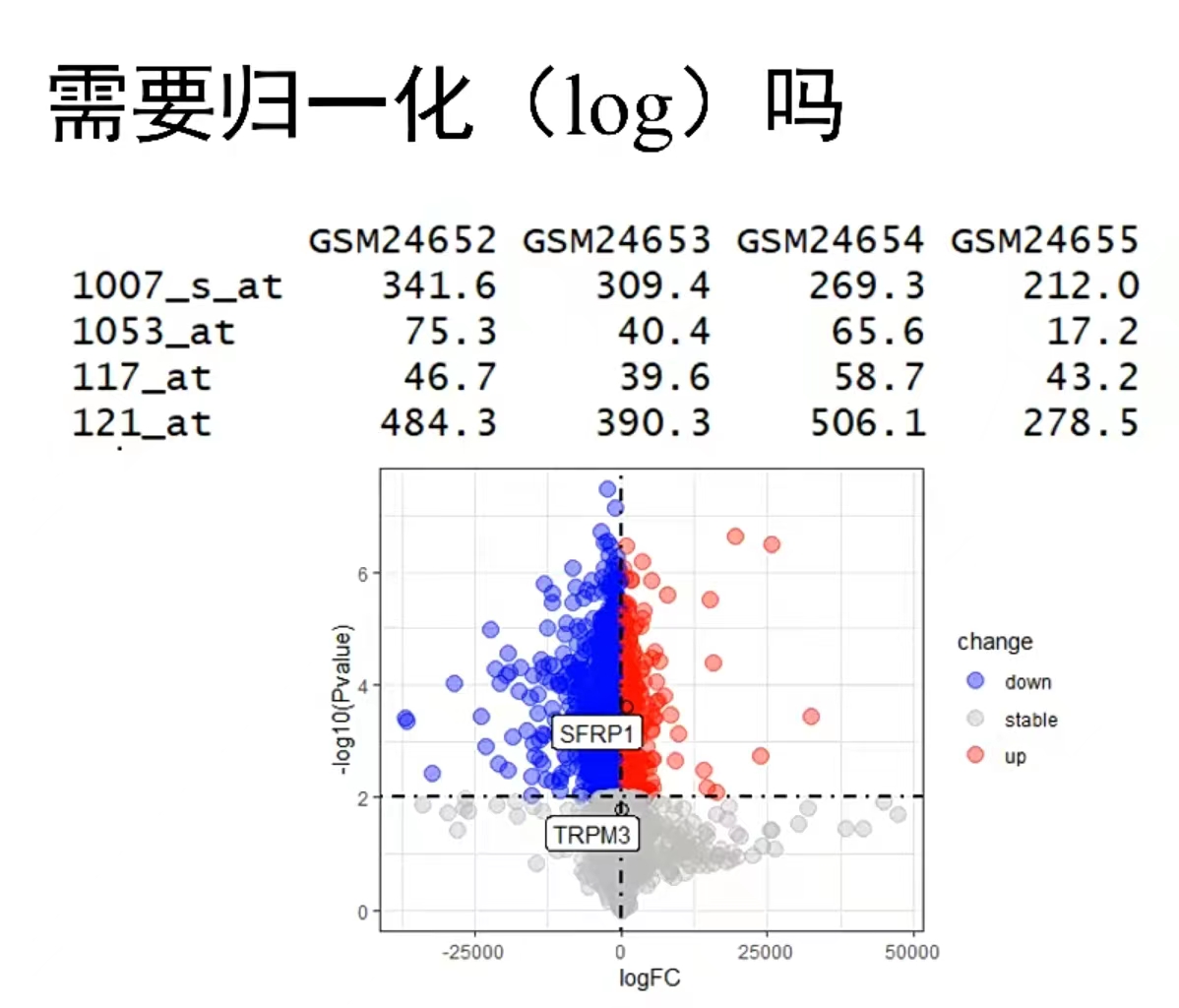
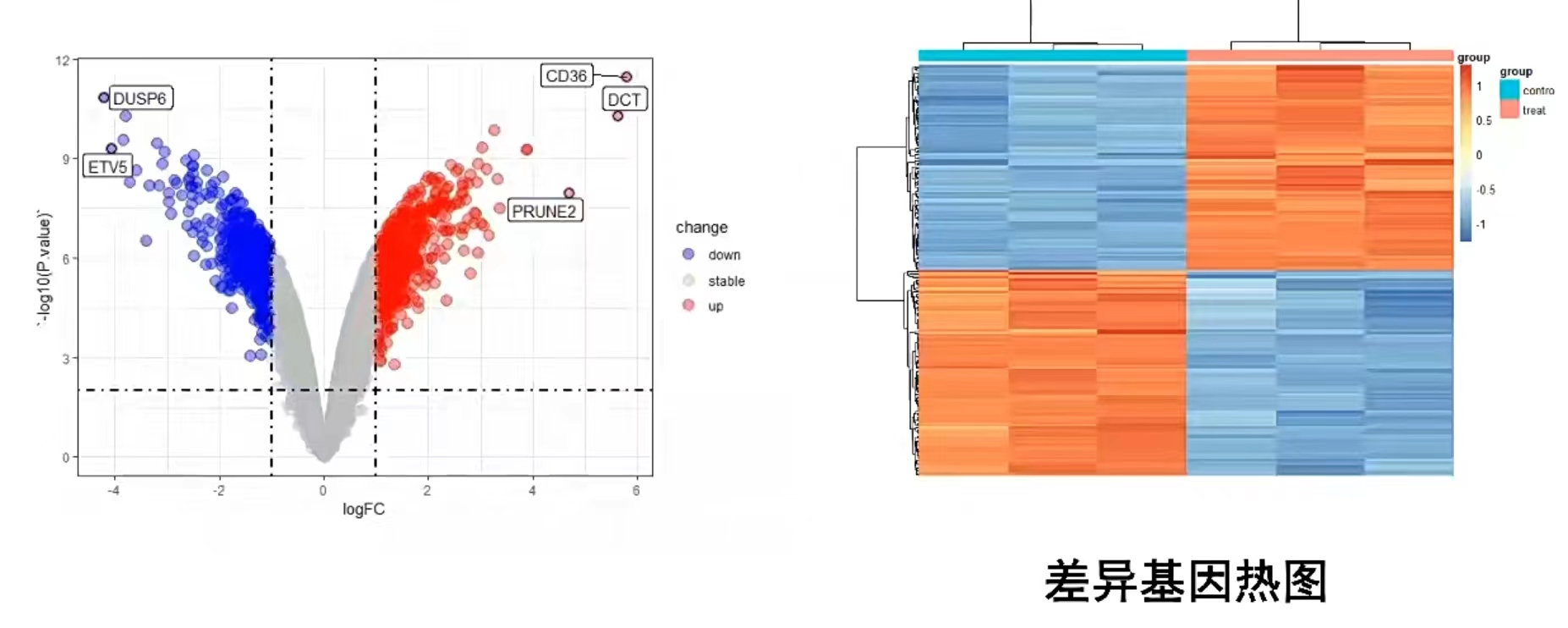
4. 差异分析及可视化,火山图、热图
limma包用于芯片差异分析
本质上只是R包和函数!
准备好需要输入的数据、写对参数
帮助文档
(另外,转录组数据差异分析三大R包:limma(voom)、edgeR、Deseq2)
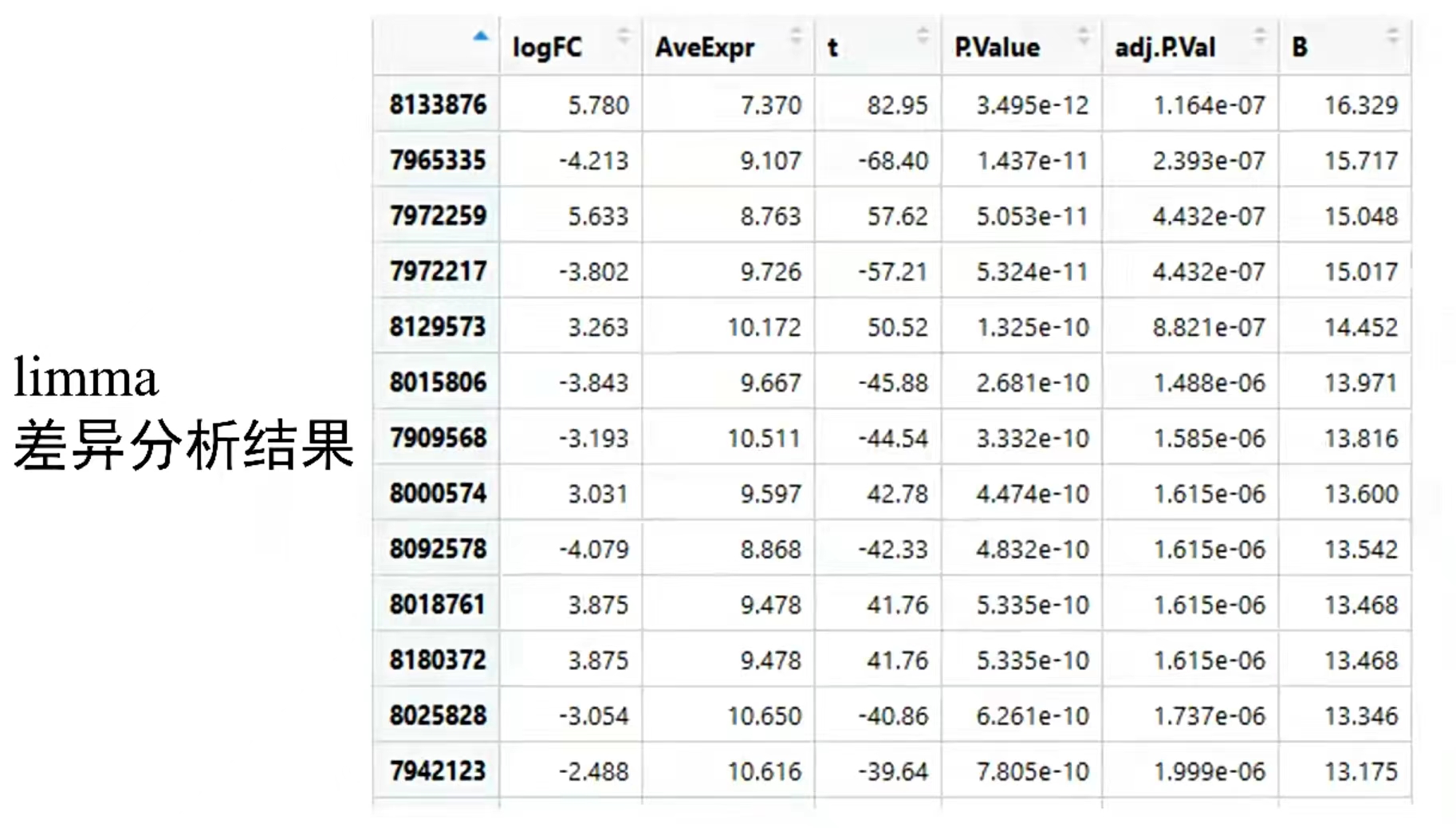
limma差异分析的结果
行名:探针的ID,继承于表达矩阵的行名。
每一行都计算了logFC和P.Value
芯片注释:探针与基因的对应关系
每个公司的芯片命名方式各不相同
基因名也有很多种
注释来源:
① GEO数据库中GPL页面的表格;
② Biocoductor的注释包 ( http://www.bio-info-trainee.com/1399.html );
③官网下载对应产品的注释表格(一般不用)
④自主注释(一般不用,因为需要比较好的R语言基础和比较大的服务器)
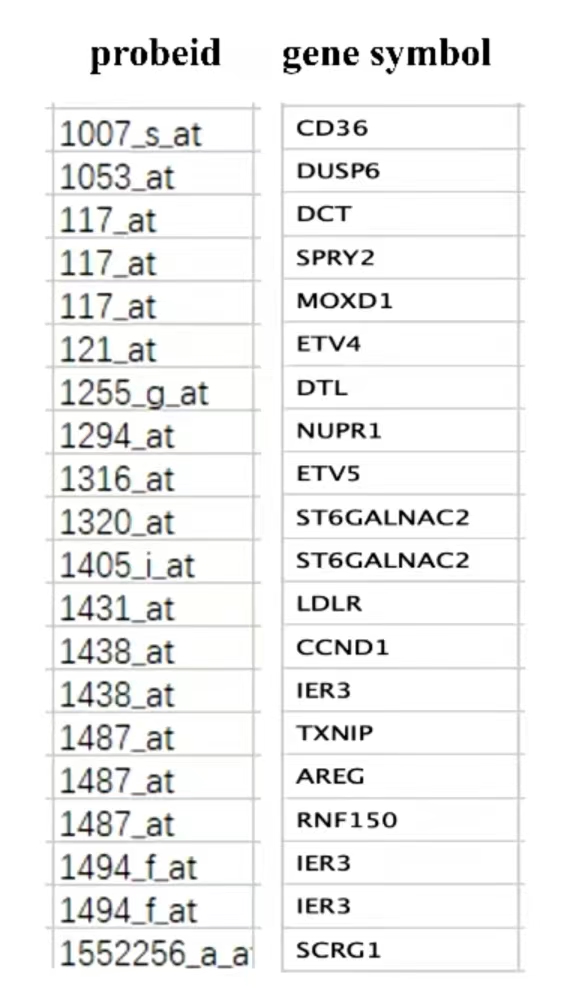
一个探针对应多个基因 - 非特异性探针,注释文件中去除,不需要管
多个探针对应一个基因:按照基因去重复
5. 富集分析
输入数据:
差异基因的entrezid,所有基因的entrezid。
id转换:
bitr()
Symbol:基因名
ENTREZID:富集分析指定用
二者并非一一对应,损失部分基因属于正常。

输出结果:
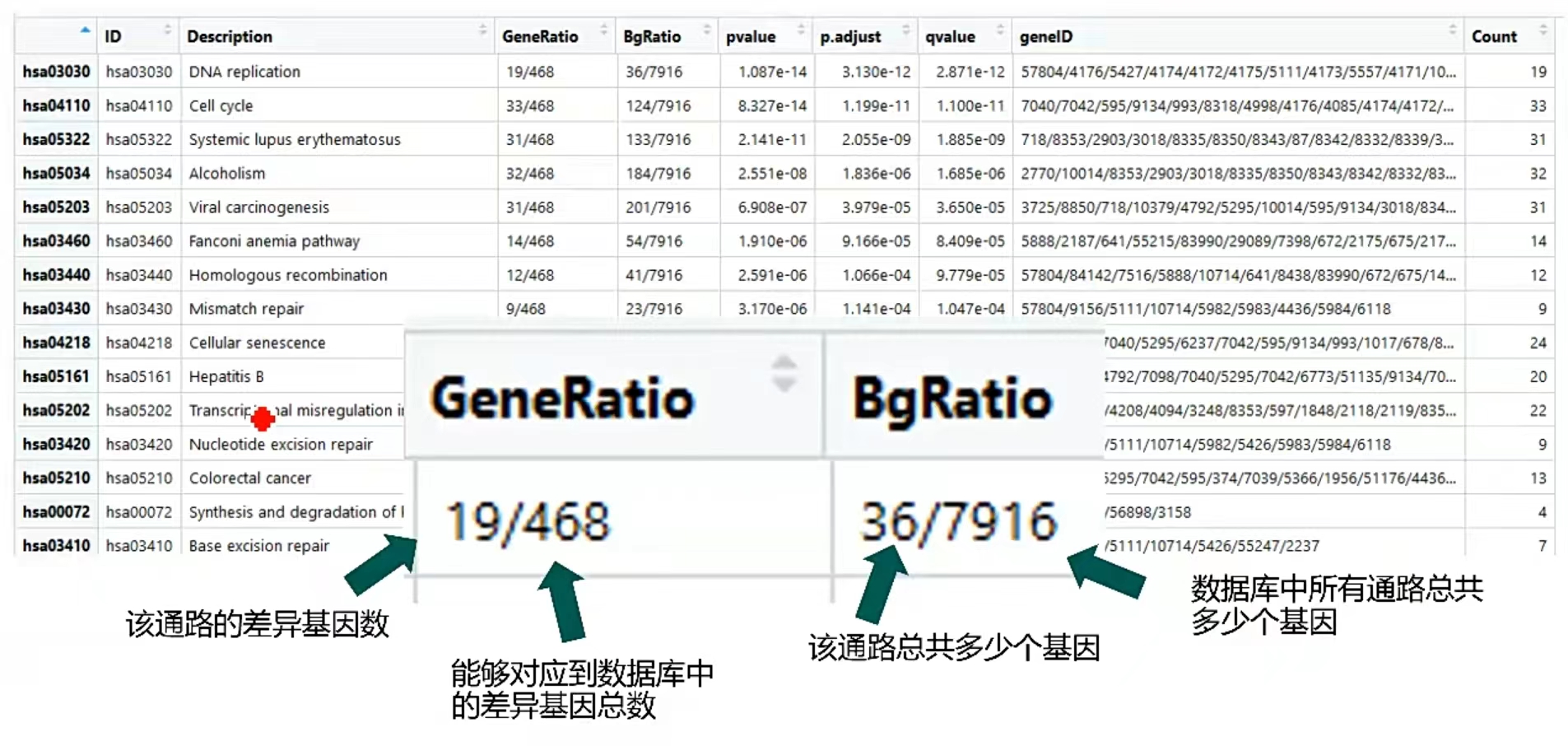
输出结果的可视化:
条带图:
横坐标:每条通路富集到的count数
纵坐标:通路名称,按p值从大到小排序
颜色:按adjust.pvalue值从大到小分配
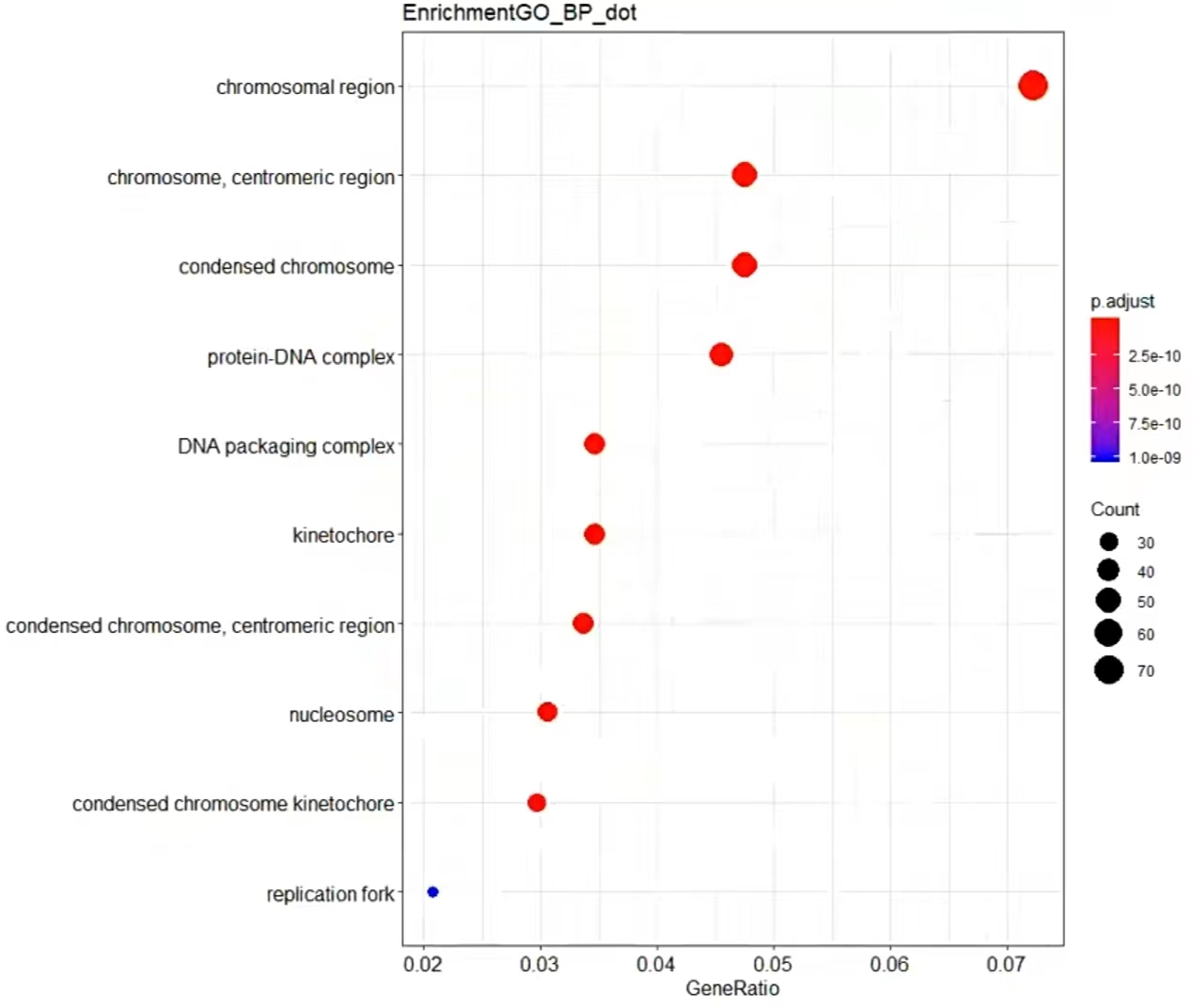
点图(气泡图):按富集基因数从大到小排列
横坐标:GeneRatio
纵坐标:通路名称,按照count数排序
颜色:根据P值
大小:根据count数
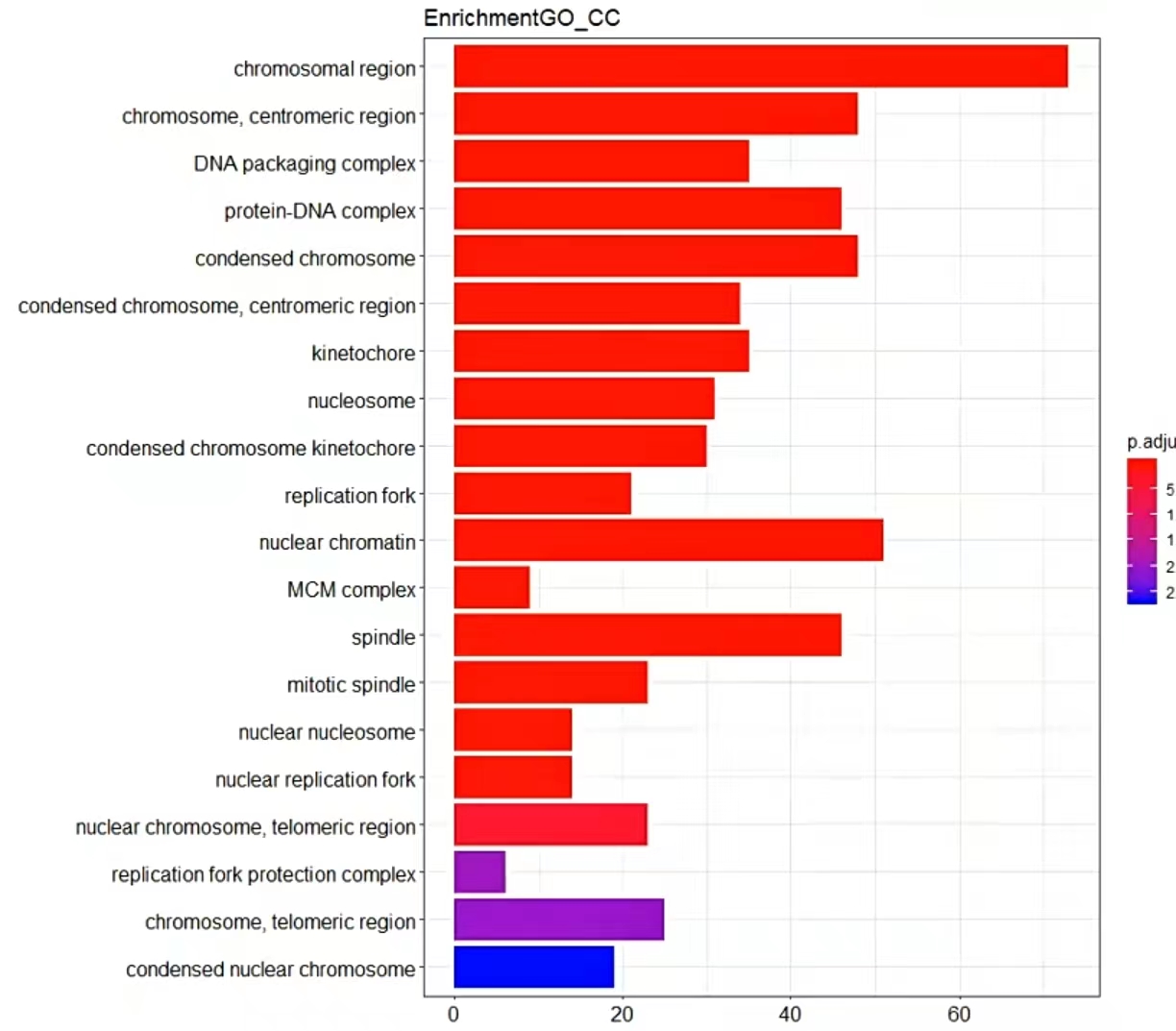
其它图
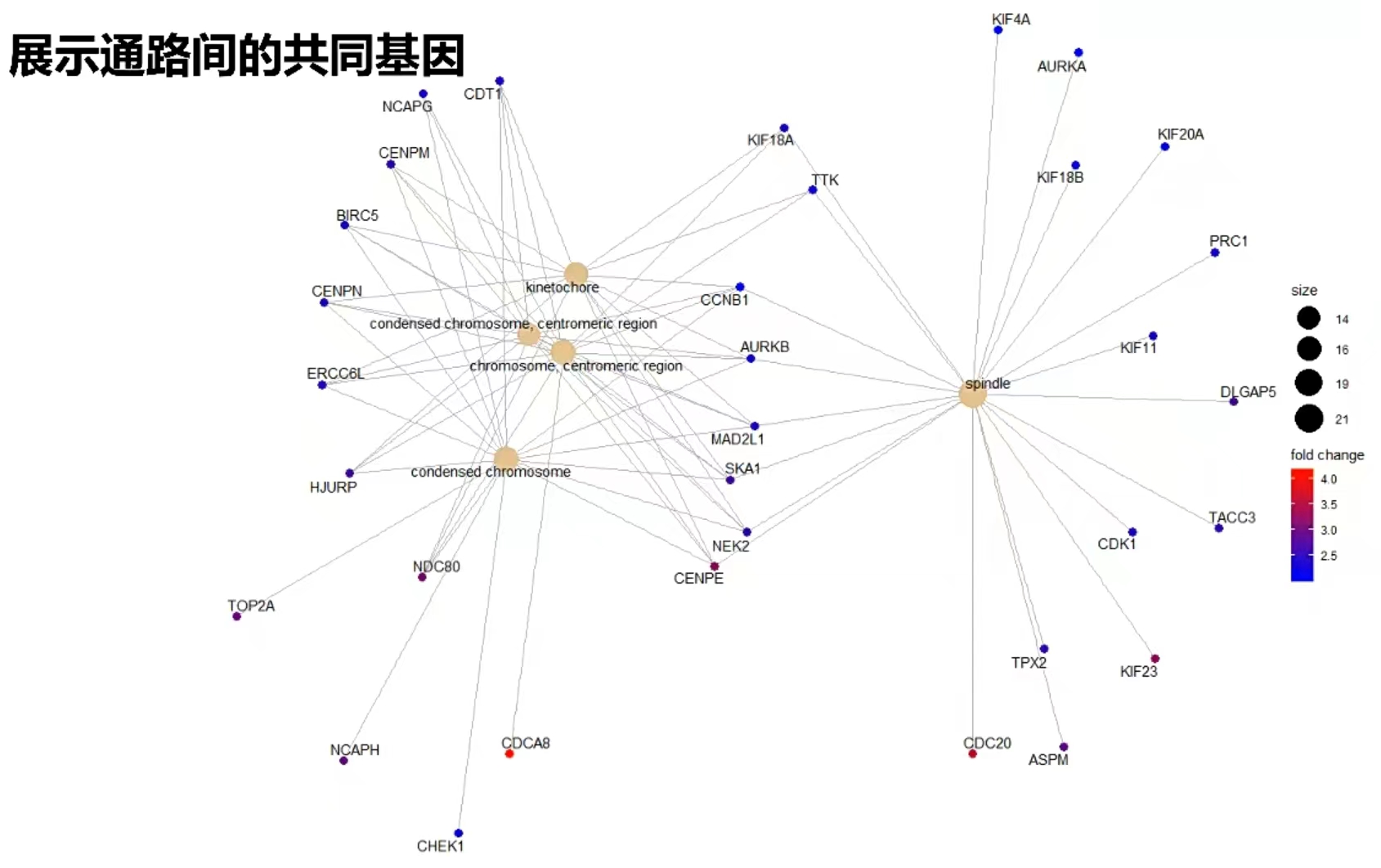
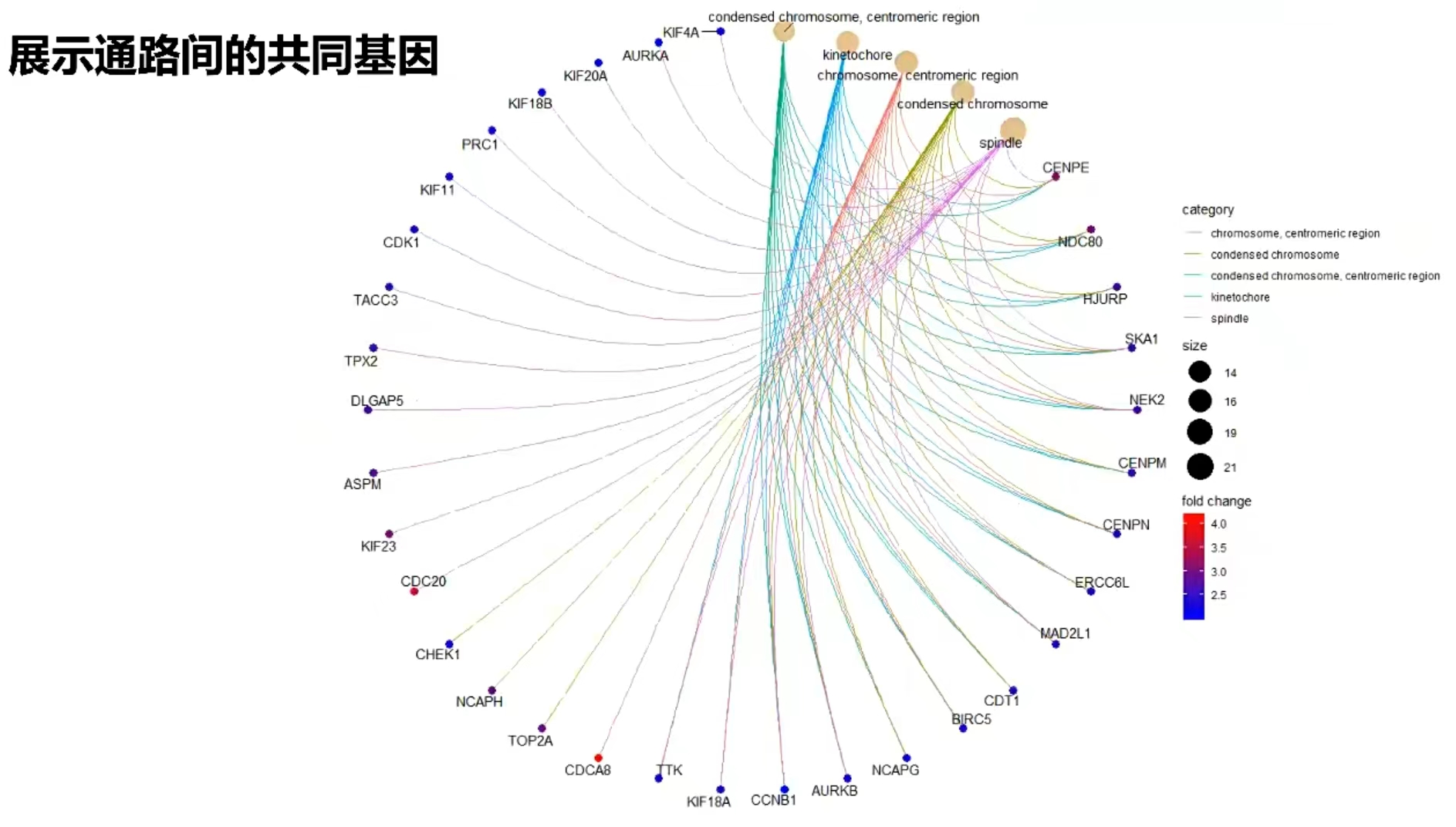

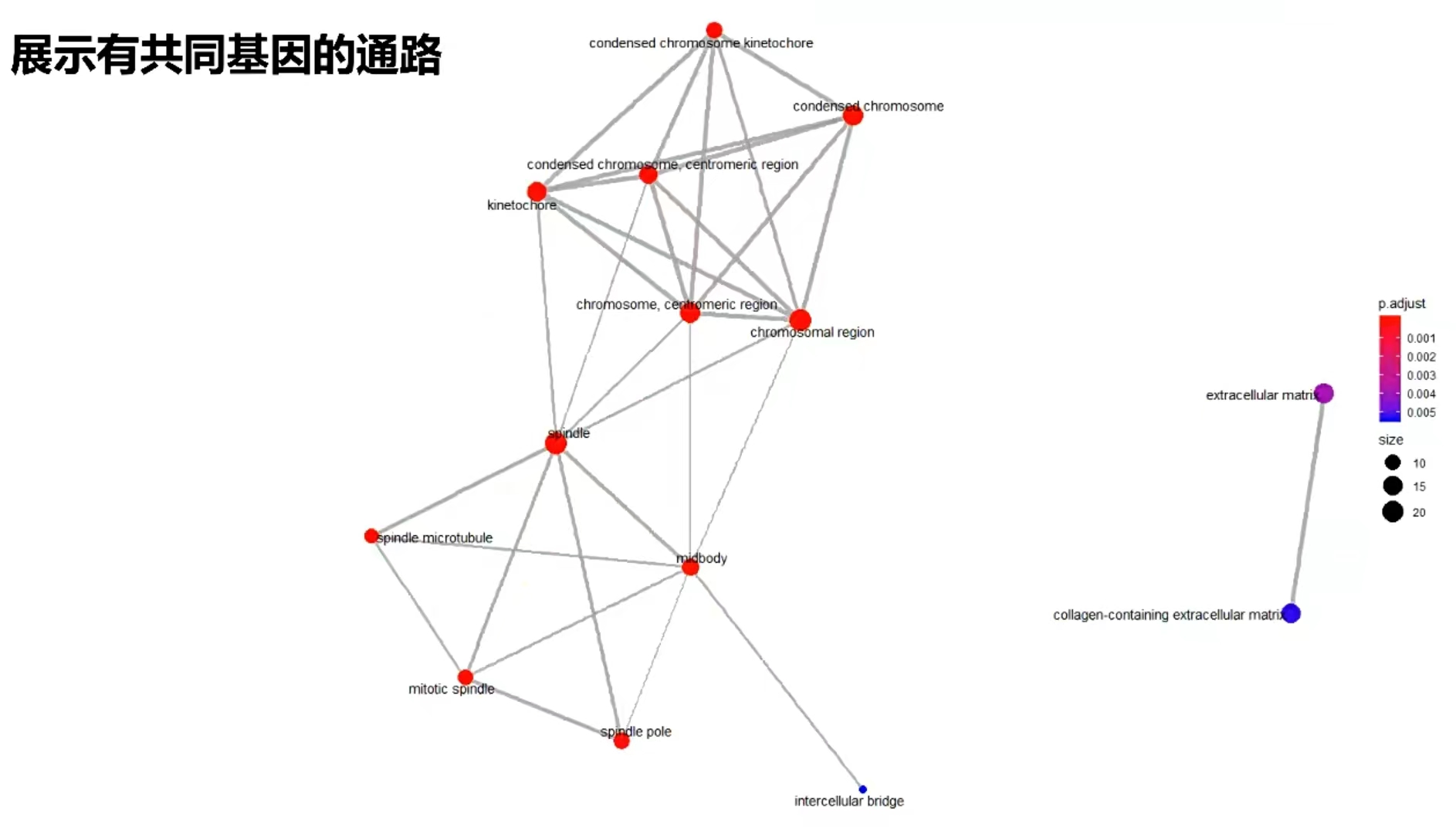
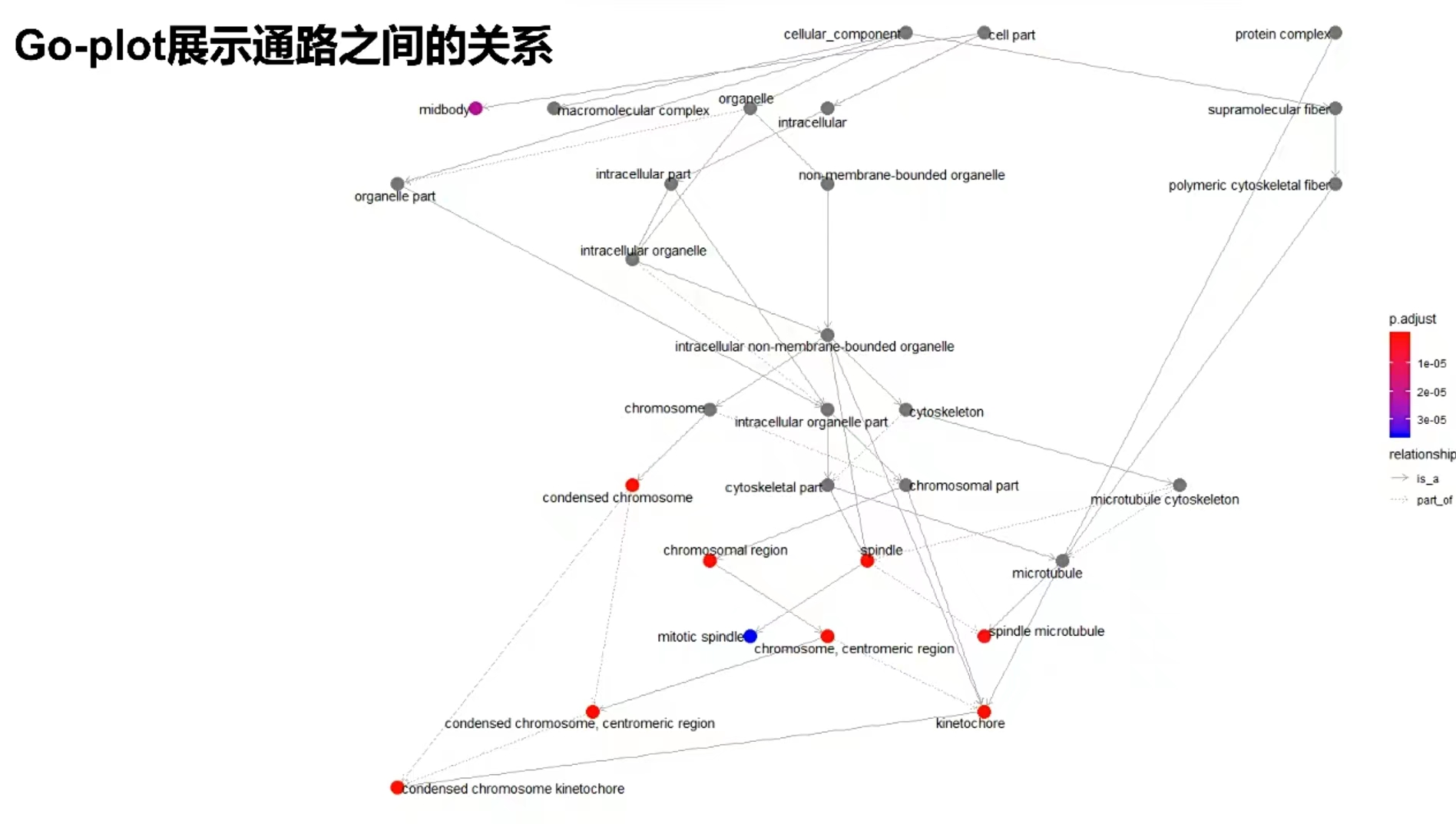

 浙公网安备 33010602011771号
浙公网安备 33010602011771号