残基和原子
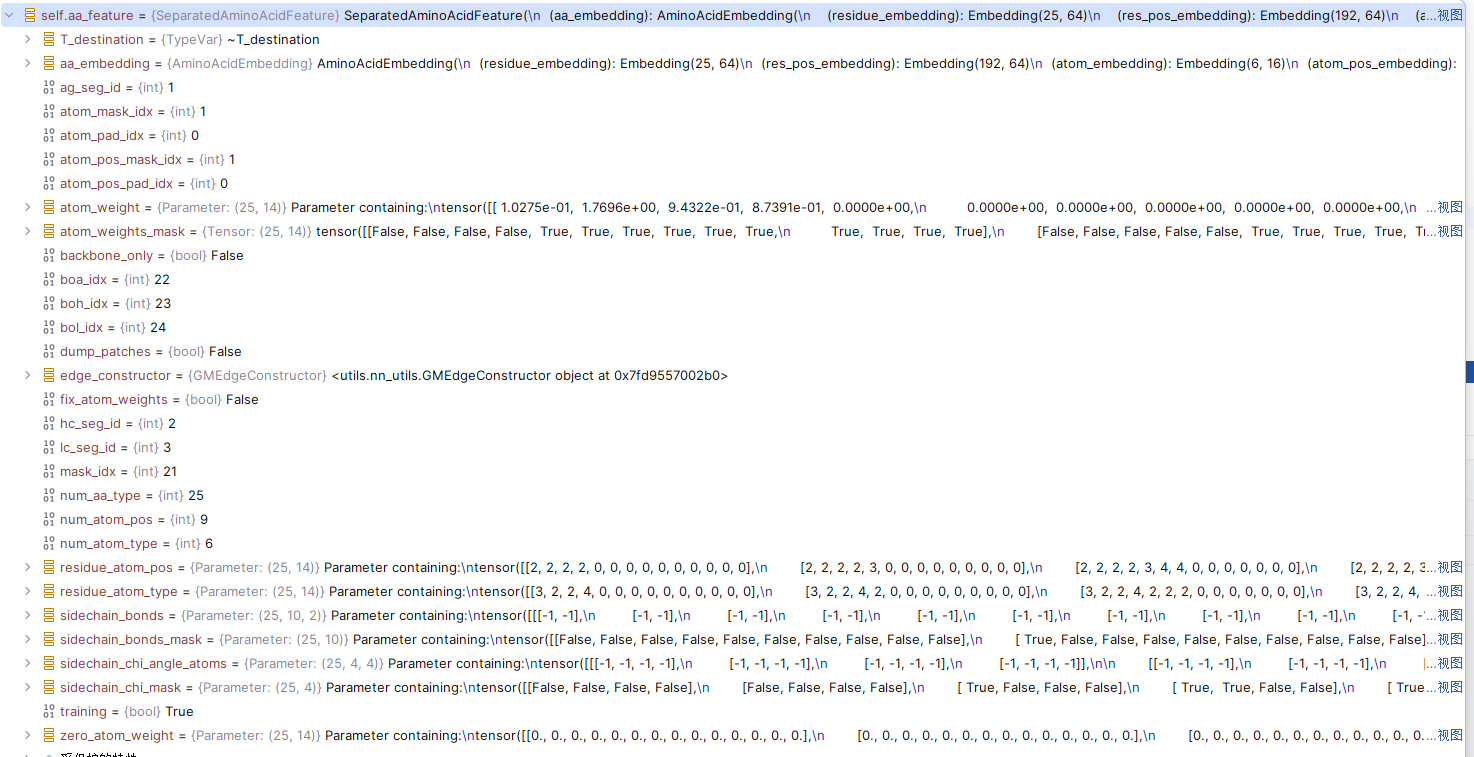
从您提供的 aa_feature 类的截图信息来看,以下是对 aa_feature 类中各个属性的整理:
主要属性说明
-
aa_embedding:
residue_embedding: 一个嵌入层,形状为 (25, 64),用于表示氨基酸残基的嵌入。res_pos_embedding: 一个嵌入层,形状为 (192, 64),用于表示氨基酸残基的位置嵌入。atom_embedding: 一个嵌入层,形状为 (6, 16),用于表示原子类型的嵌入。atom_pos_embedding: 一个嵌入层,形状为 (9, 16),用于表示原子位置的嵌入。
-
atom_pad_idx: 值为 0,表示填充原子的索引。
-
atom_mask_idx: 值为 1,表示掩码原子的索引。
-
atom_pos_mask_idx: 值为 1,表示原子位置掩码的索引。
-
atom_pos_bb_idx: 值为 2,表示骨架原子的索引。
-
num_aa_type: 值为 25,表示氨基酸的种类数量(通常是20种标准氨基酸 + 特殊标记)。
其他属性
- boa_idx, boh_idx, bol_idx: 分别为 22, 23, 24,可能是用于表示不同链(抗原链、重链、轻链)起始位置的索引。
- mask_idx: 值为 21,表示掩码索引。
- num_atom_type: 值为 6,表示原子类型的数量。
- num_atom_pos: 值为 9,表示原子位置的数量。
- dump_patches: 一个布尔值,默认
False,是否用于某些调试或保存中间结果。 - backbone_only: 一个布尔值,默认
False,表示是否仅包含骨架原子。 - fix_atom_weights: 一个布尔值,表示是否固定原子权重。
关于边和侧链的相关参数
- sidechain_bonds: 一个张量,表示氨基酸侧链原子的键连接关系。
- sidechain_bonds_mask: 一个布尔张量,表示侧链键的掩码。
- sidechain_chi_angles_atoms: 包含 χ 角信息的参数,用于表示侧链旋转构象。
其他
- edge_constructor: 可能是一个与图结构相关的对象,用于构建图中的边连接关系。
- residue_atom_pos, residue_atom_type: 表示残基原子位置和类型的参数,分别记录氨基酸残基中原子的位置和类型信息。
- residue_atom_weight: 存储每个氨基酸的原子权重信息。
总结
aa_feature 类是用于对氨基酸、原子、侧链等信息进行嵌入和编码的类,属性涉及到氨基酸残基、原子位置、原子类型、侧链连接等各种信息,适用于将氨基酸序列转化为模型可处理的嵌入向量,同时考虑了骨架、侧链结构以及不同氨基酸的特征。
def get_atom_weights(self, residue_types):
weights = torch.where(
self.atom_weights_mask,
self.zero_atom_weight,
self.atom_weight
) # [num_aa_classes, max_atom_number(n_channel)]
if not self.fix_atom_weights:
weights = F.normalize(weights, dim=-1)
return weights[residue_types] 我想知道这里对于每个residue,如果它的max channel是14,但实际上它没达到max channel,那么它的atom_weights
在这个代码片段中,get_atom_weights 函数计算了每个氨基酸残基的原子权重 (atom_weights)。由于每个氨基酸的原子数量不同,实际的原子数量通常小于 max_atom_number(即 14)。在这种情况下,未达到最大通道数的部分将通过 atom_weights_mask 被填充为 0。
代码中的关键部分解释
-
self.atom_weights_mask:- 这是一个布尔张量,形状为
[num_aa_classes, max_atom_number]。其中每个元素为True表示当前氨基酸在该位置上没有原子,False表示该位置有原子。
- 这是一个布尔张量,形状为
-
self.zero_atom_weight:- 这是一个全零的权重张量,与
atom_weights的形状相同。当self.atom_weights_mask为True时,原子权重将被设置为 0。
- 这是一个全零的权重张量,与
-
weights = torch.where(self.atom_weights_mask, self.zero_atom_weight, self.atom_weight):- 这个操作会将
self.atom_weights_mask中为True的位置填充为 0,表示该位置没有原子,从而确保这些位置不影响计算。
- 这个操作会将
-
F.normalize(weights, dim=-1):- 如果
fix_atom_weights为False,则对每个氨基酸的权重向量进行归一化,确保它们在通道维度上(-1 维)具有单位范数。这意味着每个氨基酸的实际原子权重将在它的非零位置上进行归一化。
- 如果
当实际原子数量小于 max_atom_number 时,atom_weights 的情况
- 对于每个氨基酸,实际存在的原子位置会有权重值,而超出该氨基酸原子数量的部分(没有实际原子的通道位置)会被
self.atom_weights_mask设置为 0。最终结果是,atom_weights中没有实际原子的通道部分的权重值始终为 0。 - 如果
fix_atom_weights为False,实际存在的原子权重会被归一化。
举例说明
假设一个氨基酸残基的实际原子数是 6,而 max_atom_number = 14,那么对应的 atom_weights 在计算后会是一个长度为 14 的向量,其中前 6 个元素包含归一化后的权重值,后 8 个元素全为 0。
总结
对于每个氨基酸残基,如果它的实际原子数量小于 max_atom_number(14),那么 get_atom_weights 函数会将多余的通道位置的权重设为 0,确保只有实际存在的原子通道具有有效的权重值。如果 fix_atom_weights 为 False,这些有效权重还会进行归一化处理。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号