易基因|染色质免疫共沉淀测序(ChIP-seq)分析实验全流程
本期,易基因小编给您讲讲染色质免疫共沉淀测序(ChIP-seq)实验怎么做,从技术原理、建库测序流程、信息分析流程和实验成功的关键问题等四方面详细介绍。
一、染色质免疫共沉淀测序(ChIP-seq)技术原理
蛋白质与DNA相互识别是基因转录调控的关键,也是启动基因转录的前提。ChIP技术是在全基因组范围内检测DNA与蛋白质体内相互作用的一种标准方法[1],该技术由Orlando等[2]于1997年创立,最初用于组蛋白修饰的研究,后来被广泛应用到转录因子作用位点的研究中[3]。
染色质免疫共沉淀-高通量测序(ChIP-Seq):是指通过染色质免疫共沉淀技术(ChIP)特异性地富集与目的蛋白结合的DNA片段,并对其进行纯化和文库构建;然后对富集得到的DNA片段进行高通量测序,是目前在全基因组水平研究蛋白结合靶DNA序列的重要手段,为转录因子、组蛋白修饰、核小体定位等表观遗传学的研究提供有效方法。
二、染色质免疫共沉淀测序(ChIP-seq)技术流程
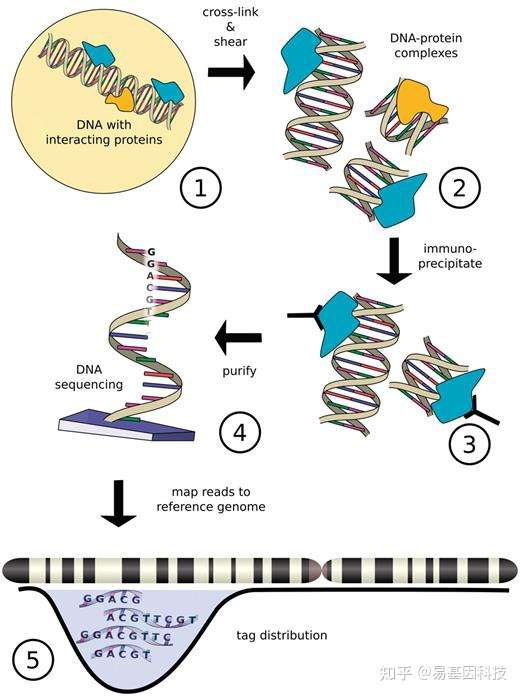
三、染色质免疫共沉淀(ChIP-seq)信息分析流程
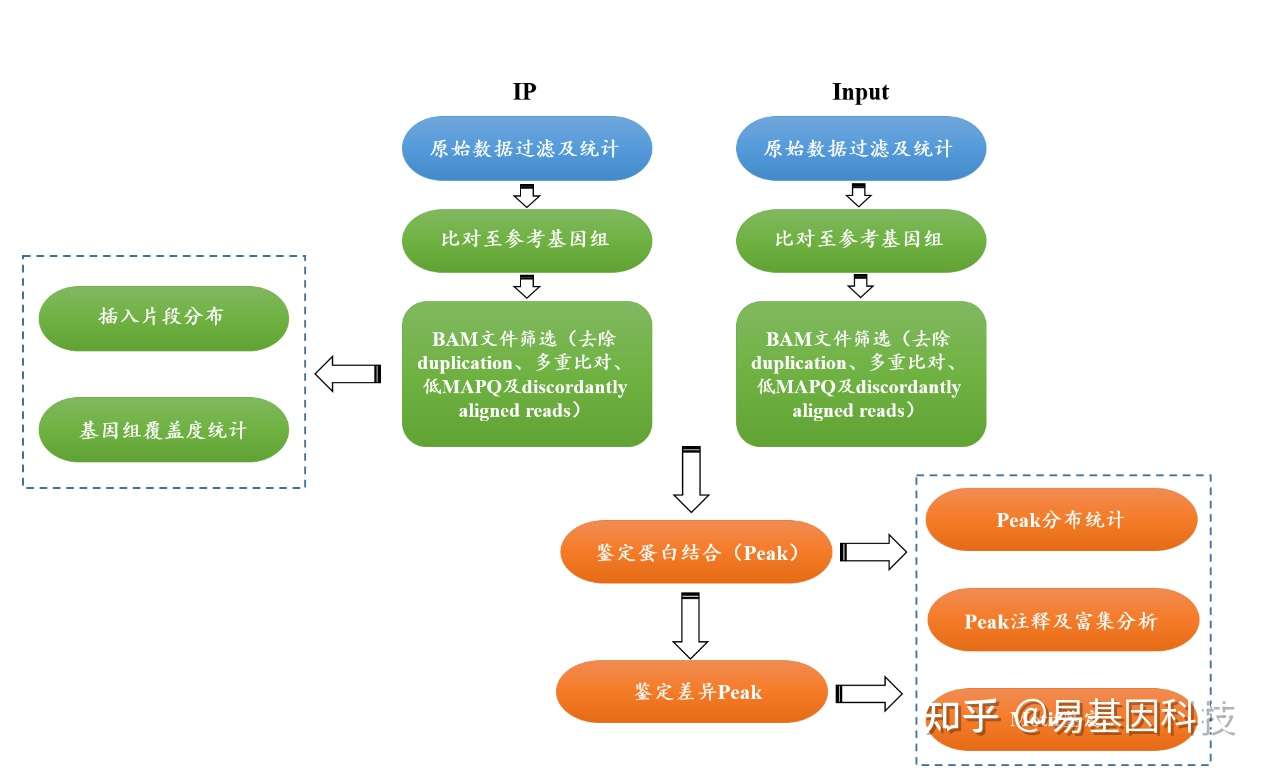
(一)原始下机数据质控
原始下机数据为FASTQ格式,是高通量测序的标准格式。FASTQ文件每四行为一个单位,包含一条测序序列(read)的信息。该单位第一行为read的ID,一般以@符号开头;第二行为测序的序列,也就是read的序列;第三行一般是一个+号,或者与第一行的信息相同;第四行是碱基质量值,是对第二行序列的碱基的准确性的描述,一个碱基会对应一个碱基质量值,所以这一行和第二行的长度相同。以下为一条read信息的示例:
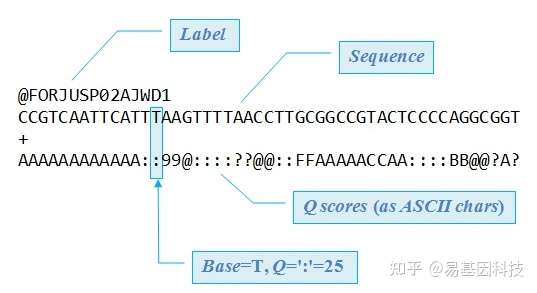
原始下机数据包含建库时引进的接头序列以及质量过低的碱基,这些因素会导致后续比对到基因组的reads较少,从而导致得到的信息较少,因此需要进行过滤。
(二)数据比对
过滤后的数据需要比对至参考基因组,基因组上与目标蛋白结合的DNA片段会有大量reads比对上去,从而形成“峰”(peak),根据峰的位置即可判断基因组的哪些区域与目标蛋白进行列结合。
如比对采用bowtie2,该软件可快速将短序列比对至参考基因组。比对完成后,对结果文件进行以下过滤:
(1)去除duplication。由于PCR过度扩增,会导致同一个模板DNA在文库中出现多次,因此可能被多次测到,这种片段称为duplication。这些片段的存在不仅不会增加有用信息量,反而会导致后续统计产生偏差,因此需要去除。
(2)去除多重比对reads。多重比对reads指比对至基因组多于一个位置的reads,这些reads的存在可能影响统计结果的准确性。
(3)去除MAPQ<30的reads。MAPQ表示该read的比对质量,其计算方法为:
Q = -10 log10 p,其中p为该read比对错误的概率。MAPQ值越高,表示该read比对的正确率越高。
(三)组蛋白修饰/蛋白结合位点的鉴定及统计
ChIP-seq将DNA上的组蛋白修饰/蛋白结合区域富集后进行测序,因此组蛋白修饰/蛋白结合区域,IP文库所覆盖的reads数会显著高于Input文库,从而形成“峰(peak)”。检测这些峰的位置即可得到DNA上组蛋白修饰/蛋白结合的区域(peak)。如采用软件MACS2[4]检测峰。
鉴定得到peak后,再利用R包ChIPseeker[5]对得到的peak进行注释、分布统计。再利用R包clusterprofiler[6]对注释基因进行GO[7]富集分析,利用KEGG等数据库进行pathway富集分析。
(四)差异组蛋白修饰/蛋白结合位点的鉴定及统计
鉴定差异组蛋白修饰/蛋白结合位点也就是鉴定差异peak。如利用MACS2的bdgdiff鉴定差异peak。该软件根据一定的规则将两样本(无生物学重复)的peak进行比较,根据一定标准筛选,得到统计学显著的差异peak。
鉴定得到差异peak后,再利用R包ChIPseeker对得到的差异peak进行注释、分布统计,利用软件HOMER进行motif鉴定等分析。
四、染色质免疫共沉淀测序实验成功的关键问题
(1)抗体质量
ChIP-seq是基于抗体的免疫沉淀实验,因此它的数据质量好坏直接取决于抗体的质量和特异性。
另外,针对同一蛋白的不同抗体,可能会识别不同的表位(尤其是单克隆抗体)。因此建议针对同一感兴趣蛋白测试不同的抗体,通过Western blot检测knock-down前后的差异帮助选择。
(2) 测序数据量
为了捕获所有真实的结合位点,而我们看不见摸不着,只能通过测序的reads去计算来帮助判断,因此测序reads的数量是一个决定因素。
需要多少reads呢?
这个取决于基因组的大小和感兴趣因子的结合方式(sharp regions for TFs and broad regions for histone marks)。哺乳动物中,鉴定TFs至少要满足10-20M,broad histone marks至少要10-45M,input对照要和ChIP样本保持同样测序深度。reads数量还取决于抗体质量和免疫沉淀的效率。信噪比越高,需要的reads数可以适当减少。
(3)生物学重复
样本重复可以看到实验设计的好坏,选择相关性高的样本进行后续分析
推荐三个生物重复,但两个现在也能接受(最粗略的实验设计就是:每个ChIP样本2个重复,input只有一个没有重复)
如果样本间的本质差异越大,越需要设置重复,例如从不同人取的样本。
五、易基因染色质免疫共沉淀测序项目文章案例
(一)组蛋白修饰ChIP-seq
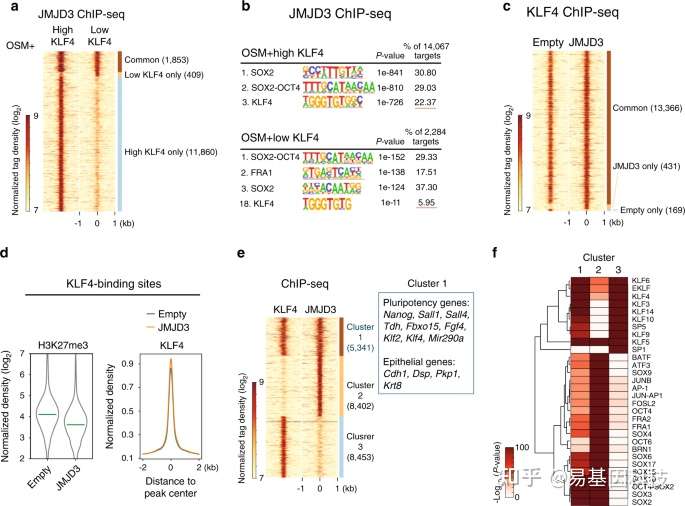
Huang Y,et al.JMJD3 acts in tandem with KLF4 to facilitate reprogramming to pluripotency. Nat Commun. 2020 Oct 8;11(1):5061. ChIP-seq揭示H3K27me3去甲基化酶在体细胞重编程调控转录机制
- 背景
作为个体发育和干细胞分化中最重要的组蛋白修饰之一,H3K27me3标记发育分化基因并抑制其表达,在基因组水平H3K27me3的动态变化是发育和分化得以有序进行的重要基础。体细胞重编程是发育和分化的逆向过程,H3K27me3势必经历逆向的时空变化。
- 方法
将小鼠饲养在病原体的环境中,12小时光照/黑暗循环,温度保持22-24°C,相对湿度40–70%,吸入二氧化碳进行安乐死,提取样本对体细胞重编程,进行RNA-seq、ATAC-seq和ChIP-seq等测序分析。
- 结论
作者通过对小鼠体细胞重编程过程中进行转录组和ChIP-seq等测序分析,揭示了H3K27me3去甲基化酶JMJD3与KLF4在体细胞重编程中协同调控转录新机制。首先,JMJD3对重编程有2方面相反的作用;在机制上,JMJD3被KLF4特异性地招募至上皮和多能性基因位点,并辅助KLF4激活这些基因。进一步,作者还在多种其他KLF4介导的细胞命运转变中验证了JMJD3的这一作用模式。
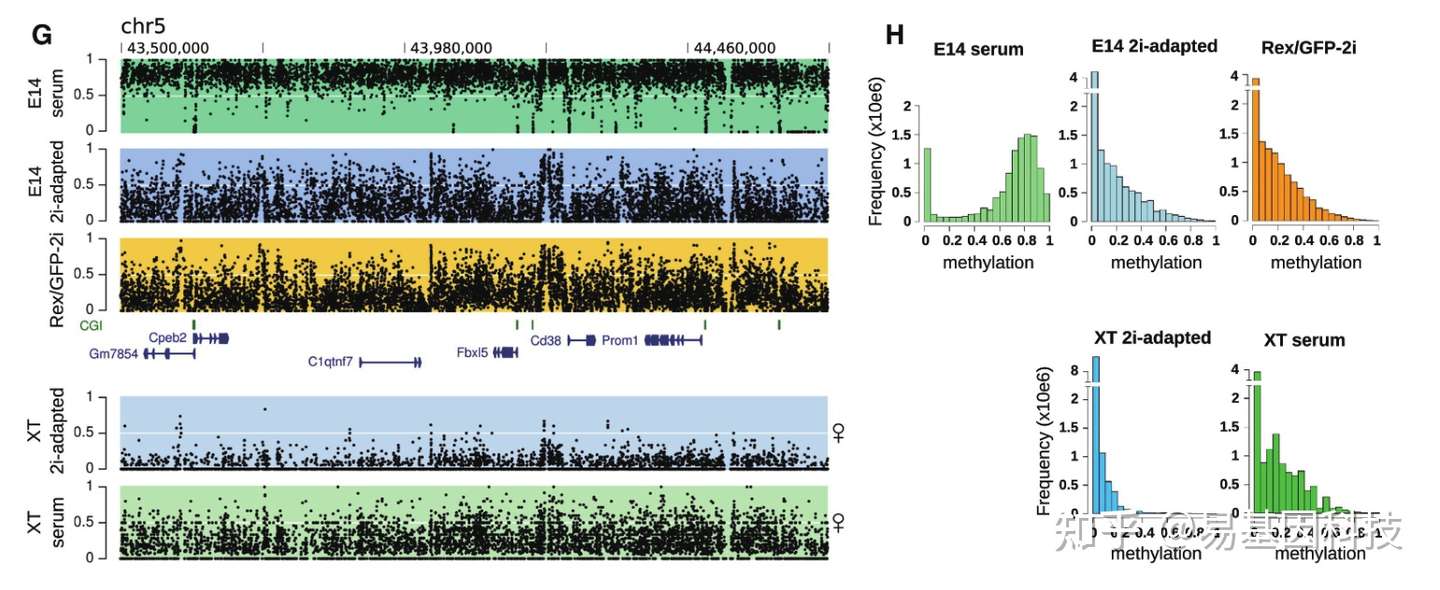
(二)转录因子ChIP-seq
Schlafen 5 suppresses human immunodeficiency virus type 1 transcription by commandeering cellular epigenetic machinery. ChIP-seq揭示HIV-1感染细胞转录抑制因子Schlafen 5的表观遗传调控机制
- 背景
Schlafen-5(SLFN5)是一种干扰素诱导的Schlafen家族蛋白,参与免疫反应和肿瘤发生。然而目前对其抗HIV-1(human immunodeficiency virus type 1 transcription)功能知之甚少。
- 方法

- 结论
在本研究中,作者鉴定出SLFN5的过表达抑制了HIV-1的复制并降低病毒mRNA水平,而内源性SLFN5缺失则促进HIV-1复制。此外,染色质免疫沉淀 (ChIP-seq+ChIP-qPCR)检测结果表明SLFN5通过与U5-R区域的两个序列结合,显著降低HIV-1长末端重复序列(LTR)的转录活性,从而抑制RNA聚合酶II(RNA PoL II)向转录起始位点募集。诱变研究验证了核定位序列和N-末端1-570氨基酸片段在抑制HIV-1中的重要性。进一步机制研究表明,SLFN5与PRC2复合物、G9a和组蛋白H3的成分互作,从而促进H3K27me2和H3K27me3修饰,导致HIV-1转录沉默。且SLFN5阻断了潜伏期HIV-1激活。总之,本研究结果表明,SLFN5是通过表观遗传调控的HIV-1转录抑制因子,是HIV-1潜伏期的潜在决定因素。
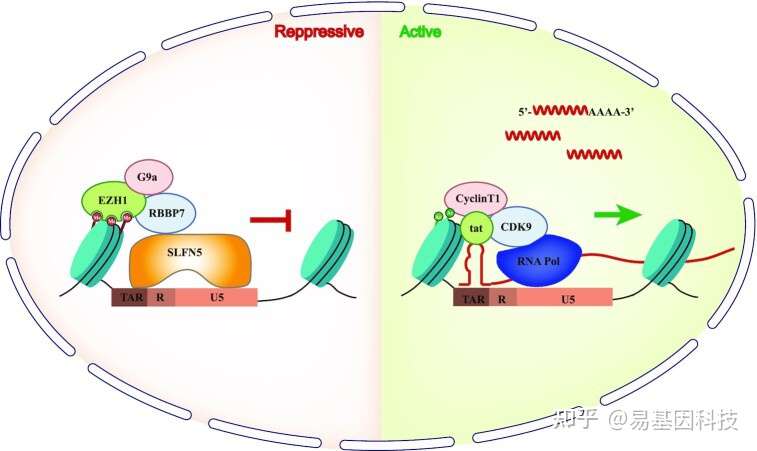
以上就是关于染色质免疫共沉淀测序(ChIP-seq)实验流程和分析思路的介绍,你学会了吗?
参考文献:
[1] Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137.
[2] Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997 Feb;11(2):205-14.
[3] Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000 Dec 8;103(6):843-52.
[4] Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. doi:10.1186/gb-2008-9-9-r137.
[5] Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015 Jul 15;31(14):2382-3.
[6] Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284-287. doi:10.1089/omi.2011.0118.
[7] Ashburner M, Ball CA,et al, Gene ontology: tool for the unification of biology. Nat Genet. 2000 May;25(1):25-9.
[8] Huang Y,et al.JMJD3 acts in tandem with KLF4 to facilitate reprogramming to pluripotency. Nat Commun. 2020 Oct 8;11(1):5061.
[9] Jiwei Ding, et al. Schlafen 5 suppresses human immunodeficiency virus type 1 transcription by commandeering cellular epigenetic machinery, Nucleic Acids Research, Volume 50, Issue 11, 24 June 2022, Pages 6137–6153.
想要了解更多关于ChIP-seq相关知识的老师,可以留言私信~后期我们会写出来分享哦~

 浙公网安备 33010602011771号
浙公网安备 33010602011771号