

组装三代番木瓜基因组——by Serenity Fang
# 估算测序深度、reads数目、N50等值(自写perl程序):
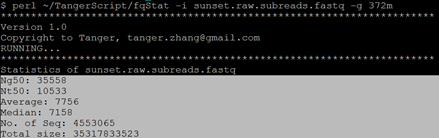
$ perl ~/TangerScript/fqStat -i sunset.raw.subreads.fastq -g 372m
统计结果如下:
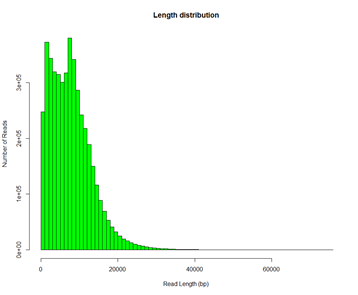
# 基因组组装三步走1. Correction 2. Assembly 3. Polish
## Step1: canu组装(1. Correction 2. Assembly)
$ (nohup) canu -s spec.txt -p sunset -d sunset-auto genomeSize=400m -pacbio-raw sunset.raw.subreads.fastq &
$ cat spec.txt 注:spec文件为配置文件,根据不同服务器设置不同的参数。

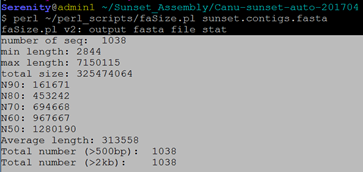
### 组装初步结果如下(自写perl程序):
$ cd /public1/home/Serenity/Sunset_Assembly/Canu-sunset-auto-201704
$ perl ~/perl_scripts/faSize.pl sunset.contigs.fasta
### 抽取unassembled.fasta中reads>5的contigs(自写python程序)
$ python ~/python_scripts/extract_faread_filter.py sunset.unassembled.fasta
### 将上一步结果与 sunset.contigs.fasta合并
$ cat sunset.contigs.fasta sunset.unassembled.fastareadfilter > sunset.all.contigs.fasta
## Step2: 第一轮矫正(3. Polish): quiver——取至少50x的三代数据做校正
$ cd /public1/home/Serenity/Sunset_Assembly/Canu-sunset-auto-201704/canu-quiver
$ ln -s ../sunset.all.contigs.fasta .
$ perl ~/TangerScript/runQuiver.pl -i sunset.all.contigs.fasta -d /public4/zhangxt/DATA/Papaya/sunset/baxh5 -t 16 注:run Quiver矫正,-t 设置节点数16-24个
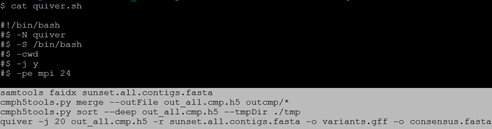
$ for i in {1..27};do qsub script/script.${i}.pbs; done 注:结束后检查outcmp里面的文件数目,检查无误后提交quiver.sh脚本
$ qsub quiver.sh 注:结束后得到consensus.fasta文件便是quiver校正后的基因组文件
## Step3: 第二轮矫正(3. Polish): pilon——取至少50x的二代数据做校正
$ cd /public1/home/Serenity/Sunset_Assembly/sunset-reseq-raw-data
### 首先统计read长度、read数目、总碱基数
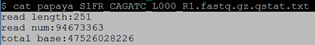
$ zcat papaya_S1FR_CAGATC_L000_R1.fastq.gz | awk 'NR==2{a=length($1)}END{print "read length:"a"\nread num:"NR/4"\ntotal base:"a*NR/4*2"\n"}' > papaya_S1FR_CAGATC_L000_R1.fastq.gz.qstat.txt
$ cat papaya_S1FR_CAGATC_L000_R1.fastq.gz.qstat.txt 注:测序深度=total base/372000000
### bwa mem进行align
$ bwa index -a bwtsw consensus.fasta
$ bwa mem -t 24 -R '@RG\tID:S1FR_CAGATC\tSM:S1FR_CAGATC\tPL:Illumina\tLB:lib1' consensus.fasta papaya_S1FR_CAGATC_L000_R1.fastq.gz papaya_S1FR_CAGATC_L000_R2.fastq.gz > papaya_S1FR_CAGATC_L000.sam
$ samtools view -bS papaya_S1FR_CAGATC_L000.sam > papaya_S1FR_CAGATC_L000.bam
$ samtools sort papaya_S1FR_CAGATC_L000.bam -o papaya_S1FR_CAGATC_L000.sorted.bam
$ samtools index papaya_S1FR_CAGATC_L000.sorted.bam
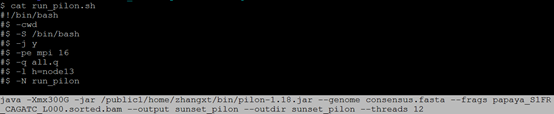
$ qsub run_pilon.sh
$ cat run_pilon.sh 注:在本实验室服务器指定13节点或者14节点,因为这两个节点内存比较大,java设置内存300G,线程设置12以上
### 组装最终结果如下:
$ perl ~/perl_scripts/faSize.pl sunset_pilon.fasta

注:N50大概达到了1.2M,总基因组大小大概组装到了330M

 浙公网安备 33010602011771号
浙公网安备 33010602011771号