
FPKM\RPKM\TPM学习[转载]
转自:http://www.360doc.com/content/18/0112/02/50153987_721216719.shtml
1.问题提出
在RNA-Seq的分析中,对基因或转录本的read counts数目进行标准化(normalization)是一个极其重要的步骤,因为落在一个基因区域内的read counts数目取决于基因长度和测序深度。
很容易理解,一个基因越长,测序深度越高,落在其内部的read counts数目就会相对越多。
当我们进行基因差异表达的分析时,往往是在多个样本中比较不同基因的表达量,如果不进行数据标准化,比较结果是没有意义的。
因此,我们需要标准化的两个关键因素就是基因长度和测序深度,常常用RPKM (Reads Per Kilobase Million), FPKM (Fragments Per Kilobase Million) 和 TPM (Transcripts Per Million)作为标准化数值。
那么,这三者计算原理是什么,有何区别呢?
2.例子说明
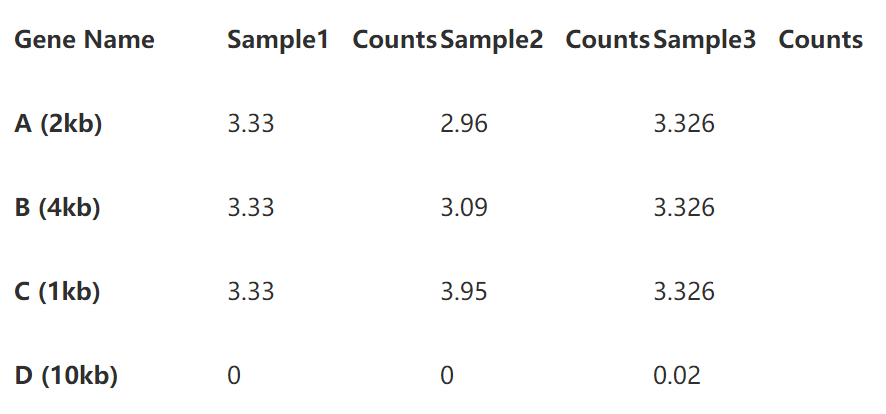
为了更清楚的展示计算过程,我们用三个样本的4个基因的read counts矩阵做例子。
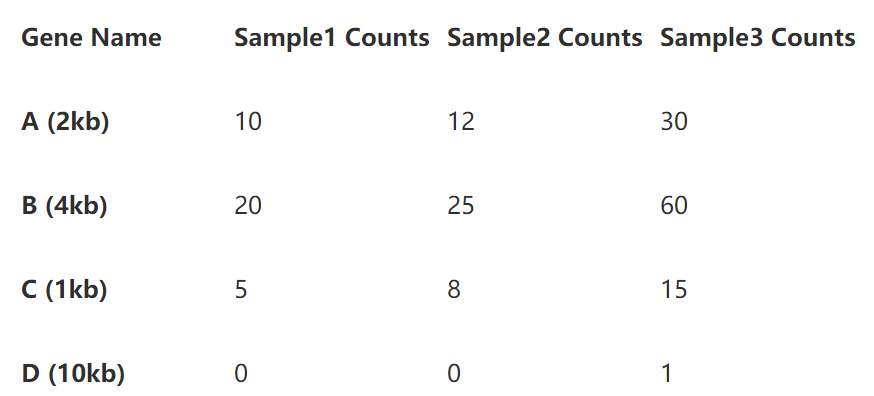
从上表,样本3的4个基因read counts数目明显多于其他两个样本,说明其测序深度较高,基因B的长度的基因A的两倍,也使得其read counts在三个样本中都高于A。
接下来我们要做就是对这个矩阵进行标准化,分别计算RPKM, FPKM和TPM, 请睁大你的眼睛(为了使数值可读性更好,下面的计算中我们用10代表million)。
2.1计算RPKM
第一步先将测序深度标准化,计算方法很简单,先分别计算出每个样本的总reads数(这里以10为单位),然后将表中数据分别除以基因长度即可,这样就得到了reads per million. 如下表:
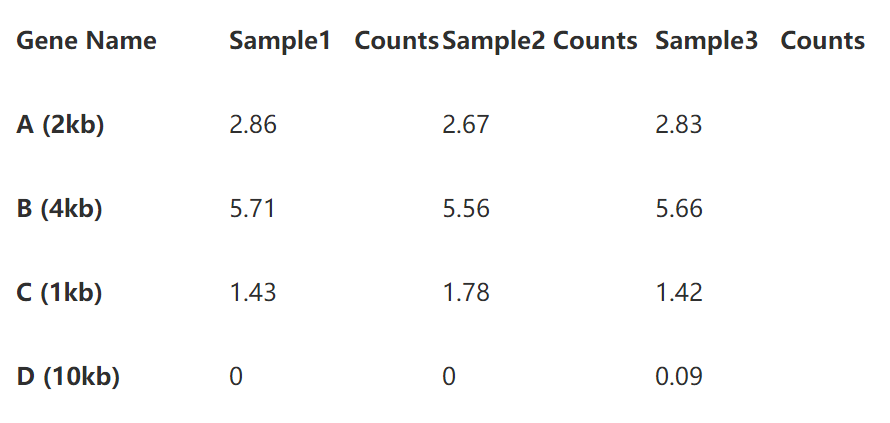
第二步即是基因长度的标准化了。将表2的read per million直接除以基因长度即可,如下表:
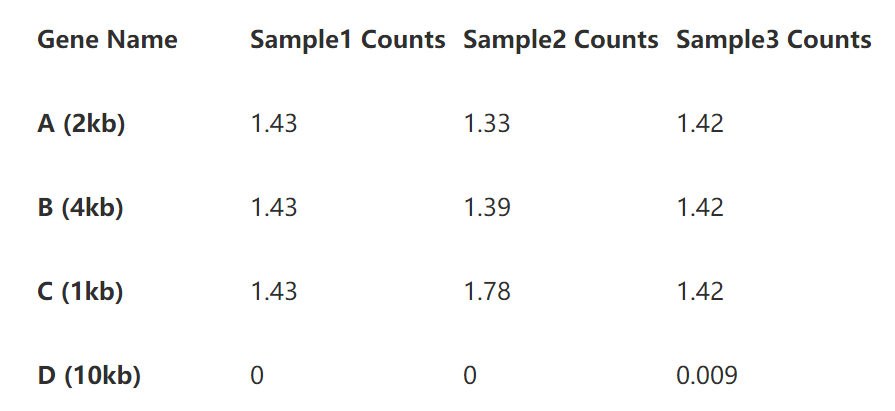
到这里,我们即得到了RPKM。
2.2FPKM
FPKM和RPKM的定义是相同的,唯一的区别是FPKM适用于双端测序文库,而RPKM适用于单端测序文库。
FPKM会将配对比对到一个片段(fragment)上的两个reads计算一次,接下来的计算过程跟RPKM一样。
在两种测序中


在FPKM中,F就是指fragment,也就是说在计算时使用的是fragment而不是reads。
那么由此可以得到在双端测序中, 如果使用RPKM计数(计数reads数目)的话,那么RPKM值将会是FPKM值的1~2倍。(呼!我终于理解了!)
2.3TPM
终于轮到TPM登场了。
虽然同样是标准化测序深度和基因长度,TPM的不同在于它的处理顺序是不同的。
即先考虑基因长度,再是测序深度。我们仍以表1的那个例子来说明TPM是计算过程。
第一步直接除以基因长度,得到reads per kilobase,如下表:
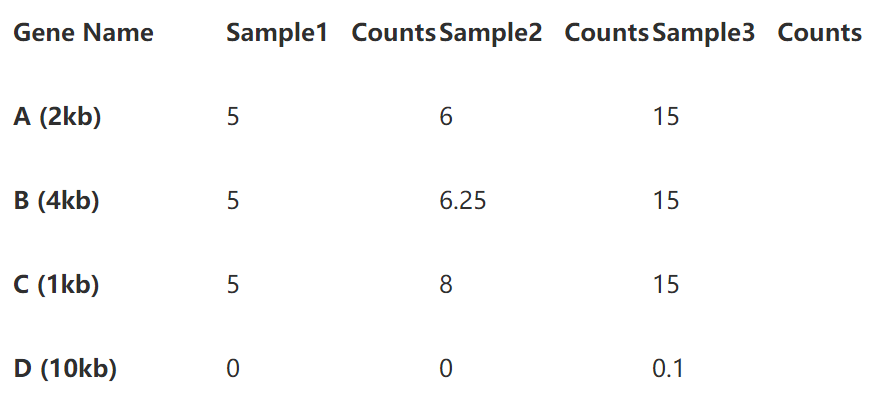
第二步标准化测序深度时,总的reads数要用第一步中除过基因长度的数值。
即第一样本除以15,第二个样本除以20.25,第三个样本除以45.1 (别忘了我们的单位是10哦)。下表就是你们想要的TPM了。
3.RPKM与TPM比较
下面,是考验你们数学功底的时候了,有没有看出来TPM分分钟完虐FPKM/RPKM?其实,只要我们在表3和表5下面多加一行你就能很轻松地看到区别了。
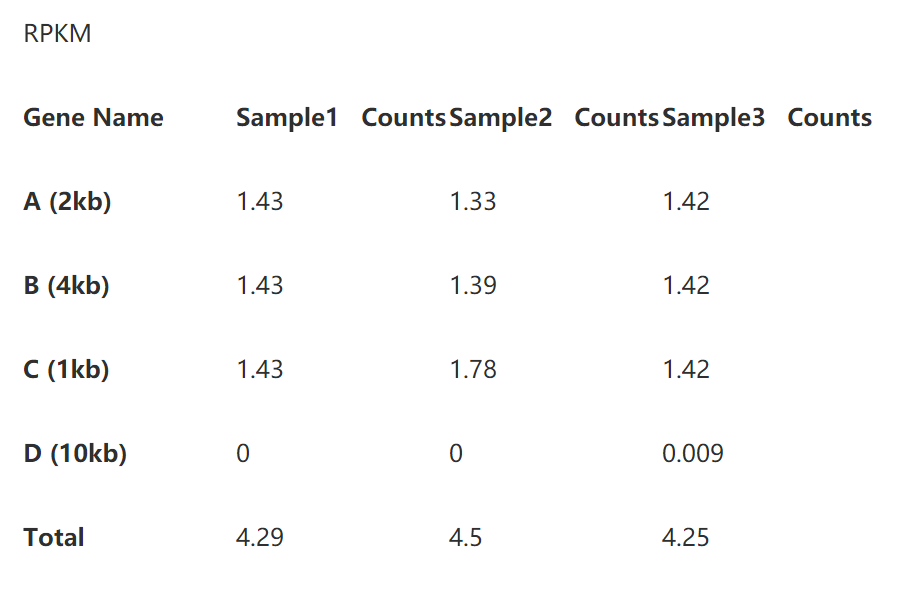
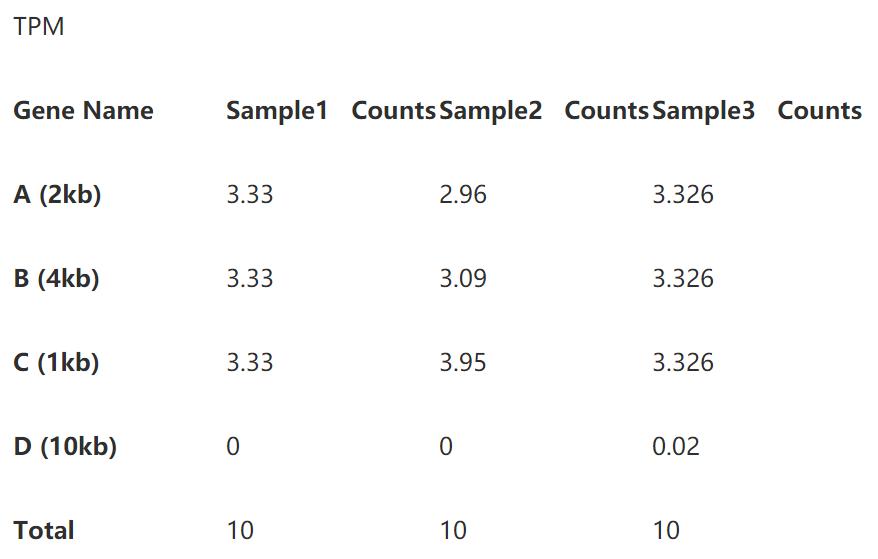
我们看到每个样本的TPM的总和是相同的,这就意味着TPM数值能体现出比对上某个基因的reads的比例,使得该数值可以直接进行样本间的比较。
//至于为什么每个sample的和是相通的,我也没有搞清楚。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号