

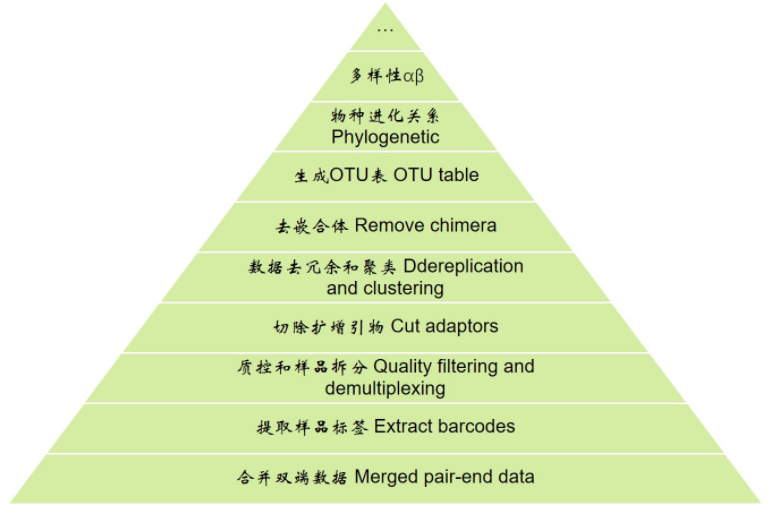
扩增子分析解读2提取barcode 质控及样品拆分 切除扩增引物
1 2 | # 进入工作目录cd example_PE250 |
1 2 3 4 5 | # 提取barcodeextract_barcodes.py -f temp/PE250_join/fastqjoin.join.fastq \ -m mappingfile.txt \ -o temp/PE250_barcode \ -c barcode_paired_stitched --bc1_len 0 --bc2_len 6 -a --rev_comp_bc2 |
barcodes.fastq # 切下来的barcode,用于后续拆分样品barcodes_not_oriented.fastq # 方向不确定序列的barcode。连引物都不匹配,质量太差,建议不再使用reads1_not_oriented.fastq # 方向不确定序列的序列,可能barcode切错方向。连引物都不匹配,质量太差,不建议使用reads2_not_oriented.fastq # 空文件reads.fastq # 序列文件,与barcode对应,用于下游分析
1 2 3 4 5 6 | # 质控及样品拆分split_libraries_fastq.py -i temp/PE250_barcode/reads.fastq \ -b temp/PE250_barcode/barcodes.fastq \ -m mappingfile.txt \ -o temp/PE250_split/ \ -q 20 --max_bad_run_length 3 --min_per_read_length_fraction 0.75 --max_barcode_errors 0 --barcode_type 6 |
histograms.txt # 所有序列长度分布数据,可知长度范围308-488,峰值为408seqs.fna # 质控并拆分后的数据,序列按样品编号为SampleID_0/1/2/3split_library_log.txt # 日志文件,有基本统计信息和每个样品的数据量;查看可知每个样品最大数据量为110454,最小值为10189
1 2 3 4 5 6 7 8 | # 下载,请尽量检查主页下载最新版源码wget https://pypi.python.org/packages/16/e3/06b45eea35359833e7c6fac824b604f1551c2fc7ba0f2bd318d8dd883eb9/cutadapt-1.14.tar.gz# 解压tar xvzf cutadapt-1.14.tar.gz# 进入程序目录cd cutadapt-1.14/# 安装在当前用户目录,不需管理员权限python setup.py install --user |
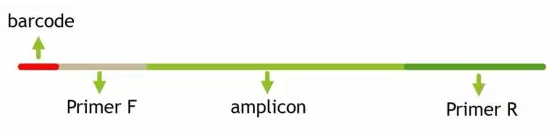
1 | cutadapt -g AACMGGATTAGATACCCKG -a GGAAGGTGGGGATGACGT -e 0.15 -m 300 --discard-untrimmed temp/PE250_split/seqs.fna -o temp/PE250_P5.fa |
This is cutadapt 1.14 with Python 3.6.1
Command line parameters: -g AACMGGATTAGATACCCKG -a GGAAGGTGGGGATGACGT -e 0.15 -m 300 --discard-untrimmed temp/PE250_split/seqs.fna -o temp/PE250_P5.fa
Trimming 2 adapters with at most 15.0% errors in single-end mode ...
Finished in 73.83 s (58 us/read; 1.04 M reads/minute).=== Summary ===
Total reads processed: 1,277,436
Reads with adapters: 1,277,194 (100.0%)
Reads that were too short: 8,849 (0.7%)
Reads written (passing filters): 1,268,345 (99.3%)Total basepairs processed: 522,379,897 bp
Total written (filtered): 495,607,409 bp (94.9%)=== Adapter 1 ===
Sequence: GGAAGGTGGGGATGACGT; Type: regular 3'; Length: 18; Trimmed: 202757 times.
No. of allowed errors:
0-5 bp: 0; 6-12 bp: 1; 13-18 bp: 2Bases preceding removed adapters:
A: 96.3%
C: 1.5%
G: 0.8%
T: 1.3%
none/other: 0.0%
WARNING:
The adapter is preceded by "A" extremely often.
The provided adapter sequence may be incomplete.
To fix the problem, add "A" to the beginning of the adapter sequence.Overview of removed sequences
length count expect max.err error counts
3 3 19959.9 0 3
4 4 4990.0 0 4
6 2 311.9 0 2
8 1 19.5 1 1
11 1 0.3 1 1
13 1 0.0 1 1
15 9 0.0 2 9
17 42 0.0 2 42
18 202626 0.0 2 202626
19 56 0.0 2 56
20 1 0.0 2 1
21 1 0.0 2 1
32 1 0.0 2 1
38 1 0.0 2 1
39 1 0.0 2 1
41 1 0.0 2 1
309 2 0.0 2 2
310 1 0.0 2 1
311 3 0.0 2 3=== Adapter 2 ===
Sequence: AACMGGATTAGATACCCKG; Type: regular 5'; Length: 19; Trimmed: 1074437 times.
No. of allowed errors:
0-5 bp: 0; 6-12 bp: 1; 13-19 bp: 2Overview of removed sequences
length count expect max.err error counts
3 2 19959.9 0 2
7 1 78.0 1 0 1
8 2 19.5 1 1 1
10 6 1.2 1 4 2
11 1 0.3 1 1
12 3 0.1 1 2 1
13 5 0.0 1 3 2
14 24 0.0 2 17 7
15 51 0.0 2 32 14 5
16 71 0.0 2 56 12 3
17 134 0.0 2 92 30 12
18 327 0.0 2 189 117 21
19 1059175 0.0 2 1056863 2069 243
20 13846 0.0 2 1817 10955 1074
21 744 0.0 2 5 10 729
22 1 0.0 2 1
23 2 0.0 2 2
24 1 0.0 2 1
25 2 0.0 2 2
27 5 0.0 2 5
28 2 0.0 2 2
29 2 0.0 2 2
30 1 0.0 2 1
31 2 0.0 2 2
32 10 0.0 2 10
49 1 0.0 2 1
51 1 0.0 2 1
166 1 0.0 2 1
291 6 0.0 2 6
401 2 0.0 2 2
409 1 0.0 2 1
443 1 0.0 2 1
460 2 0.0 2 2
465 2 0.0 2 2WARNING:
One or more of your adapter sequences may be incomplete.
Please see the detailed output above.


【推荐】还在用 ECharts 开发大屏?试试这款永久免费的开源 BI 工具!
【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· SQL Server如何跟踪自动统计信息更新?
· AI与.NET技术实操系列:使用Catalyst进行自然语言处理
· 分享一个我遇到过的“量子力学”级别的BUG。
· Linux系列:如何调试 malloc 的底层源码
· AI与.NET技术实操系列:基于图像分类模型对图像进行分类
· 对象命名为何需要避免'-er'和'-or'后缀
· JDK 24 发布,新特性解读!
· Java24你发任你发,我用Java8
· .NET Core奇技淫巧之WinForm使用Python.NET并打包
· C# 中比较实用的关键字,基础高频面试题!