
GWAS:拒绝假阳性之case和control数量比例严重失衡的解决方案(SAIGE模型的应用)
一、为什么要校正case和control数量比例不平衡情况
试问作为生信届人员,最怕的是什么,当然是统计结果不靠谱。统计结果不靠谱包括两方面:一个是假阴性,一个是假阳性。假阴性可以理解为白天鹅被误当成丑小鸭了,假阳性可以理解为一大堆青蛙,你不知道哪个才是你的真命天子。假阴性就罢了,最多让你错过发现真理的机会,但万一假阳性呢,你拿着一个看似完美的结果吭哧吭哧做实验验证,一年半载的周期下来,什么结果都验证不出来,岂不是坑了做实验的人。因此,我们就要在源头上,把这个不靠谱的统计结果杜绝出去。
上一篇文章什么!GWAS研究中case和control的比例是有讲究的?就讲到GWAS分析中,如果case和control数量比例失衡的话,会产出非常多的假阳性结果,而用SAIGE模型做GWAS分析可以校正这种数量比例不平衡的情况。下面具体讲讲怎么应用SAIGE模型。
二、怎么校正:SAIGE的下载和安装
1、下载SAIGE
此操作在Linux上进行,系统要求R-3.5.1, gcc >= 5.5.0, cmake 3.8.1
wget https://github.com/weizhouUMICH/SAIGE/blob/master/SAIGE_0.35.8.1_R_x86_64-pc-linux-gnu.tar.gz
2、安装SAIGE
R CMD INSTALL SAIGE_XX_R_x86_64-pc-linux-gnu.tar.gz
3、安装SAIGE所依赖的其他R包:Rcpp, RcppArmadillo, RcppParallel, data.table, SPAtest, RcppEigen, Matrix, methods, optparse
以下两个方法二选一:
如果是用conda的话,则用以下命令:
conda install -n r-env r-Rcpp #r-env是指conda下的R环境 conda install -n r-env r-RcppArmadillo conda install -n r-env r-RcppParallel conda install -n r-env r-SPAtest conda install -n r-env r-RcppEigen conda install -n r-env r-optparse
也可以进入R,用install.packages( )安装:
install.packages("Rcpp")
install.packages("RcppArmadillo")
install.packages("RcppParallel")
install.packages("SPAtest")
install.packages("RcppEigen")
install.packages("optparse")
三、怎么校正:SAIGE的分析、解读
1、第一步,计算NULL GLMM
1)计算NULL GLMM的命令:
Rscript step1_fitNULLGLMM.R \ --plinkFile=./input/plinkforGRM_1000samples_10kMarkers \ --phenoFile=./input/pheno_1000samples.txt \ --phenoCol=y \ --covarColList=x1,x2 \ --sampleIDColinphenoFile=IID \ --traitType=binary \ --outputPrefix=./output/example \ --nThreads=4 \ --LOCO=TRUE
plinkFile为plink的输入文件(bed, bim, fam格式)
phenoFile文件格式如下,第一列代表研究的表型,第二列及第N-1列代表协变量,最后一列IID为个体的ID号:

--phenoCol=y # 指定你要研究的表型列名,在本次例子中,指定y的表型分析。
--covarColList=x1,x2 #指定加入的协变量
--sampleIDColinphenoFile=IID #指定样本的ID
--traitType=binary #指定研究的表型的类型,binary指二分类,即case和control
--outputPrefix #生成文件的输出路径
2)输出文件的结果解读:
这个步骤会生成三个文件,分别为:example.rda、example.varianceRatio.txt、example_30markers.SAIGE.results.txt
第一个文件:example.rda,是一个model file
可以用R打开:
load("./output/example.rda")
names(modglmm)
modglmm$theta
第二个文件:example_30markers.SAIGE.results.txt,随意选取位点的关联分析结果
第三个文件:example.varianceRatio.txt
2、计算每个marker的SPA得分
1)计算每个marker的SNP得分命令:
Rscript step2_SPAtests.R \ --dosageFile=./input/dosage_10markers.txt \ --dosageFileNrowSkip=1 \ --dosageFileNcolSkip=6 \ --dosageFilecolnamesSkip=CHR,SNP,CM,POS,EFFECT_ALLELE,ALT_ALLELE \ --minMAF=0.0001 \ --sampleFile=./input/sampleIDindosage.txt \ --GMMATmodelFile=./output/example.rda \ --varianceRatioFile=./output/example.varianceRatio.txt \ --SAIGEOutputFile=./output/example.plainDosage.SAIGE.txt \ --numLinesOutput=2 \ --IsOutputAFinCaseCtrl=TRUE
dosage_10markers.txt的文件格式如下,类似于plink的ped格式,前六列分别为:CHR, SNP, CM, POS, COUNTED, ALT, 后面为个体的ID号:

sampleIDindosage.txt文件为样本ID名:

example.rda、example.varianceRatio.txt为第一步生成的两个文件。
2)输出文件的结果解读:
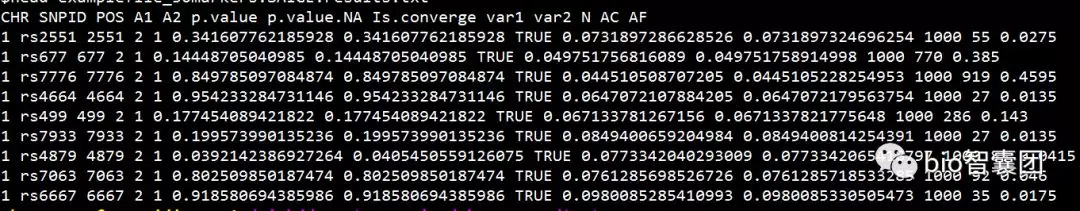
生成example.plainDosage.SAIGE.txt文件,其内容如下:

其中,P值(红框)即为我们校正case和control数量比例不平衡以后得到的GWAS结果,p.value.NA为不校正case和control数量不平衡的结果。
参数说明:
CHR: chromosome
POS: genome position
SNPID: variant ID
Allele1: Ref allele
Allele2: Alt allele
AC_Allele2: allele count of Alt allele
AF_Allele2: allele frequency of Alt allele
N: sample size
BETA: effect size
SE: standard error of BETA
Tstat: score statistic
p.value: p value with SPA applied
p.value.NA: p value when SPA is not applied
Is.SPA.converge: whether SPA is converged or not
varT: estimated variance of score statistic with sample related incorporated
varTstar: variance of score statistic without sample related incorporated
AF.Cases: allele frequency of allele 2 in cases (only for binary traits and if --IsOutputAFinCaseCtrl=TRUE)
AF.Controls: allele frequency of allele 2 in controls (only for binary traits and if --IsOutputAFinCaseCtrl=TRUE)
至此,校正GWAS分析中case和control数量比例严重失衡的工作就完成了。导师再也不用担心你给出一堆假阳性结果了。
本文来自博客园,作者:橙子牛奶糖(陈文燕),转载请注明原文链接:https://www.cnblogs.com/chenwenyan/p/10641545.html

